Pathway Editor Brief Tutorial - version 1.0 beta 2
The Pathway Editor is a program for managing, visualizing, and editing signaling and metabolic pathways. In addition to the default Pathway Editor (.path) format, the Pathway Editor can read, edit, and write files in SBML (.sbml), and BioPAX (.owl, .xml) formats. This tutorial provides an overview of Pathway Editor features, including how to start the Pathway Editor, access LIPID MAPS pathways and display LIPID MAPS and user-provided experimental data, create pathways from scratch, and edit and save pathway files and images. Instructions for working with SBML and BioPAX formats are also provided.
System RequirementsCore Features
- Download, install, and start Pathway Editor
- Open a pathway from a file
- View some selected pathway information
- Create a new pathway
- Create a node
- Show node information
- Change the node's compound
- Change the node's label
- Add more nodes
- Connect nodes to form a process
- Rearrange nodes and processes
- Change the appearance of an individual node
- Change the appearance of all nodes in a pathway or subpathway
- Change the appearance of all processes (interactions) in a pathway or subpathway
- Change the appearance of an individual process (interaction)
- Change pathway display settings
- Node label fonts
- Manage data display
- Obtain an expanded chart for printing and saving to file
- Animation
- Save a pathway as a *.path file
- Save a pathway as an image
- Using Pathway Editor to view user-defined data
- Pathway Enrichment Analysis
- Graph Modeling Language (GML) pathway layout files (.gml)
- Drawing organelles
- 3D structures in .path files
- Using compressed pathways and subpathways
- Subpathway interactions
- How to construct a layered pathway
SBML Support
BioPAX Support
- Introduction
- BioPAX model files
- Building an example BioPAX model
- BioPAX interaction participants
- BioPAX interactions
- BioPAX pathway steps
- BioPAX pathways
- BioPAX utility classes
- BioPAX options
CellML Support
Annotations
Creation of SBML, BioPAX, and CellML models from Workbench .path files
Troubleshooting
Core Features
Download, Install and Start Pathway Editor
Download and launch Pathway Editor Web Start version (no database connection; separate Java Runtime Environment required)
Download Pathway Editor installer program (includes Java Runtime Environment):
Windows (no database connection)Mac (64 bit Mac OS X version) (no database connection)
Download BioPAX Level 3 files of 25 LIPID MAPS Pathways
- created using the Pathway Editor acting on downloaded path files from our Pathway database
Click on "yes" each time you are asked if you trust our downloads (we assume you do). The Editor asks if you want to associate files ending in “.path” with this program. If you do, click "yes" on the PC, or check the box next to "Do this automatically for files like this from now on" on the Mac. This allows Pathway Editor to be placed on a list of programs that opens this file type.
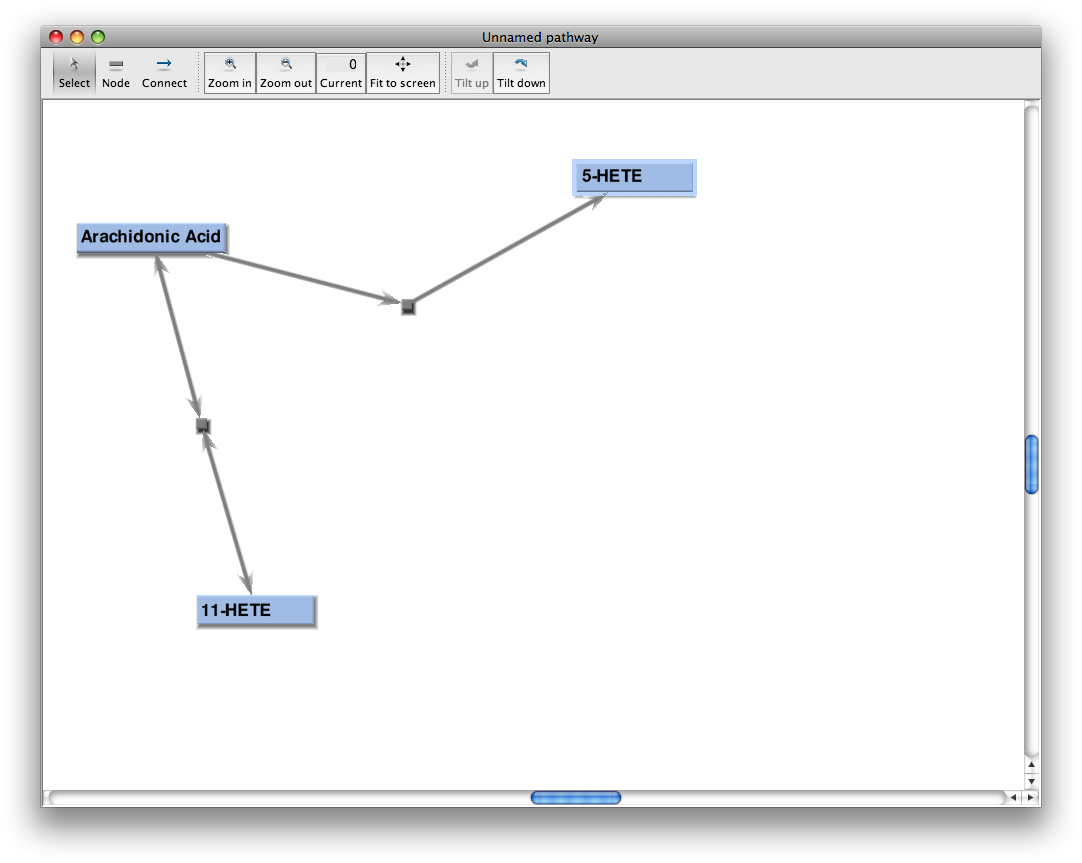
When the download completes, the editor will start up and look like this:
![]()
The 3 leftmost buttons in the toolbar at the top allow entry into node select mode, node creation mode, and node connect mode. The next two buttons allow you to zoom in and out, repectively, with the current zoom level shown in the 'Current' button. The 'Fit to screen' button fits the current pathway to the canvas. The last two buttons, 'Tilt up' and 'Tilt down', allow you to tilt the pathway plane. The scrollbars on the right and bottom of the drawing canvas allow you to move the viewing area vertically and horizontally over the drawing plane. The mouse scroll wheel allows fine-grained zooming in and zooming out in small increments.
Dialogs were designed using screen dimensions of 1920 x 1200 pixels on Windows and Macs. If you have difficulty with dialog sizes, you may wish to change your screen resolution to pixel dimensions that match these in relative proportion
Open a pathway from a file
Go to the "File" menu and select "Open a Workbench pathway file..." to get a file-opening window. Select a file, click "Open" and the pathway will load.
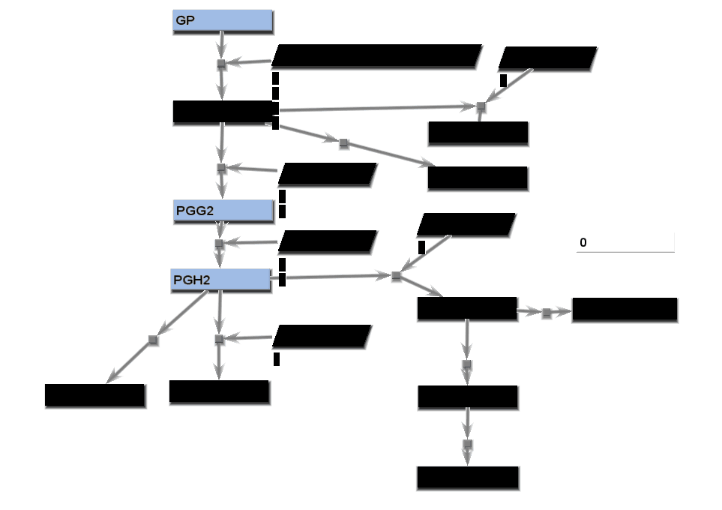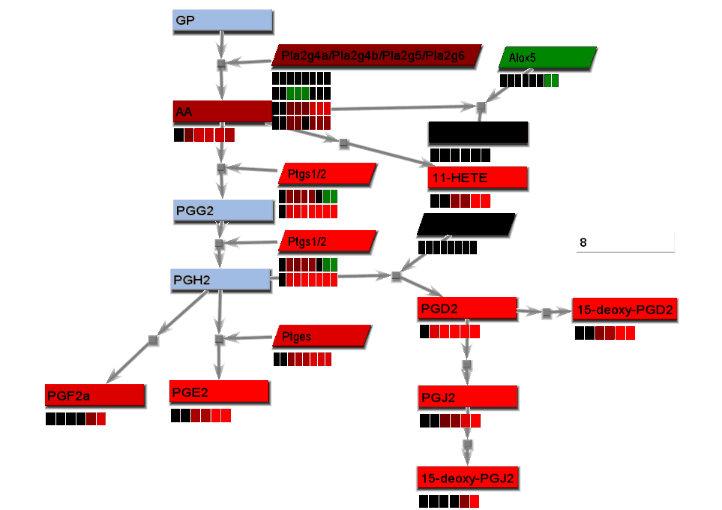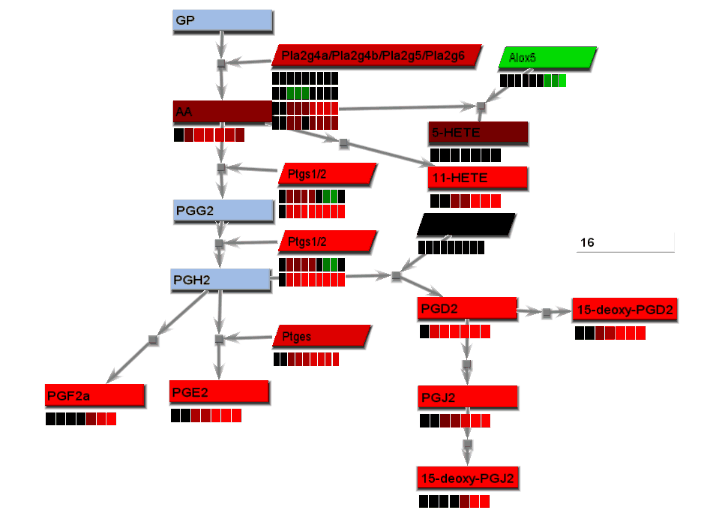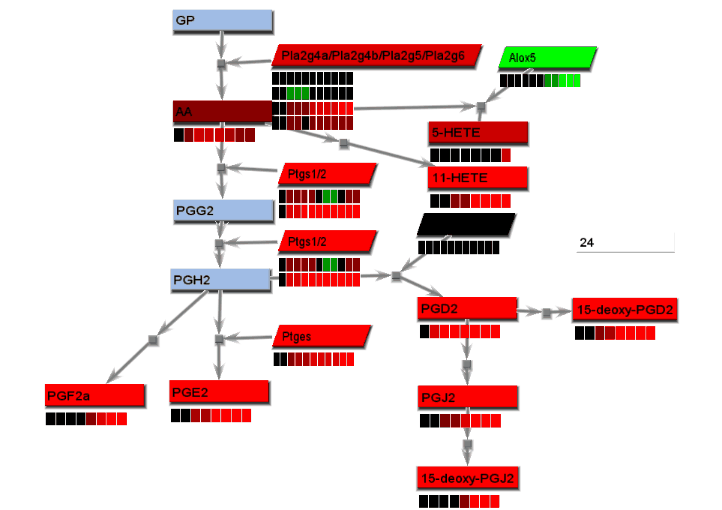
Pathway files must end with ".path". If you do not already have a .path file saved to your local environment, you can download the .path file for Arachodonic Acid metabolism: AApathway9genes.path
A .path file contains descriptions of the components of a pathway and their appearance in the Editor display. It contains database IDs (identifiers) and other material that identify specific molecules. This information is checked when the Editor opens the file and is updated from the database automatically. The file may also contain experiment IDs that reference experiment data in the LIPID MAPS database. The presence of experiment IDs instructs the Pathway Editor to download experimental data. All of the data for a time course (lipidomics) experiment is downloaded. For a microarray or protein array experiment, only the data for the microarray or protein array molecule identifiers currently being used in the file and in the currently displayed pathway are downloaded.
Additional .path files may be downloaded from http://www.lipidmaps.org/pathways/pathwayeditor.html, or accessed from within the Pathway Editor application, as described below in "Open a LIPID MAPS pathway from the database".
View some selected pathway information
Go to the "File" menu and select "Show pathway info" to get a Pathway information window.
The window contains information that may be edited and saved to a .path file. This includes pathway title, type of pathway, comments, authors, and history.
The KEGG pathway download dialog contains controls for setting viewing preferences that allow for fitting large numbers of nodes within the viewing panel. These include the zoom, size on the screen, font and node sizes, and label placement. After downloading, if you wish to perform other tasks, you might reset node and pathway preferences using dialogs from the Edit menu.
Create a new pathway
Go to the "File" menu and select "New pathway " to create a new empty pathway.
Create a node
On the toolbar, select the box-shaped icon (2nd from left) to enter node creation mode, then click in the drawing area to drop a new node at the cursor. Nodes are always box-shaped icons.
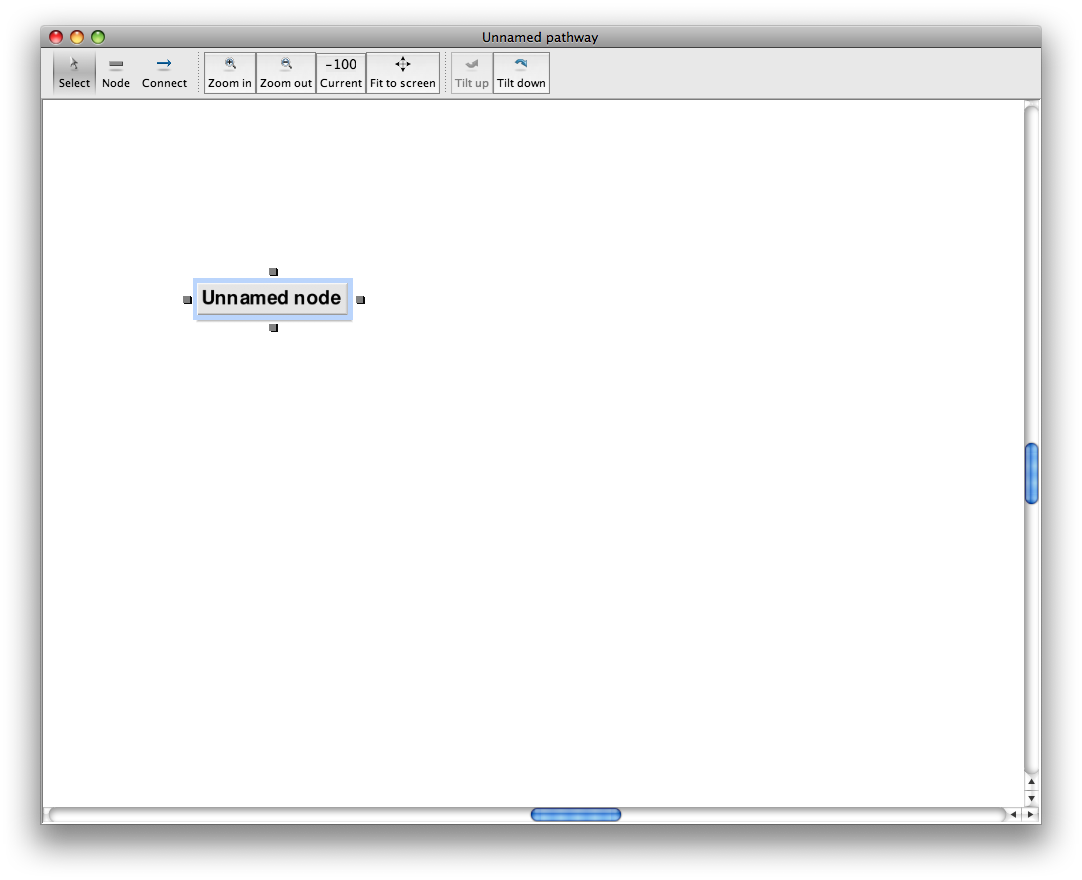
The blue outline shows that the node is selected.
Show node information
You can show information about a selected node by doing any of the following (they are all equivalent):
- Select "Show node info..." from the "Edit" menu.
- Press the right mouse button and select "Show info..." from the context menu that pops up.
- Double-click on the node or heat map/chart.
All of these bring up a window about the node. From this window you can change the node's label, compound, and other attributes.

Change the node's compound
To assign a compound to the node, select the "Compound" tab and press the "Select compound..." button. This shows a compound search window.

Constrain the search by selecting the compound type and category and/or entering a keyword. Radio buttons provide further constraints on the text that is searched. Then press "Search" to get a list of results.

Select one of the compounds from the list and press the "Select" button. The compound is loaded from the database and assigned to the node. You can view the compound's details by pressing the "Show info..." button on the node's information window.

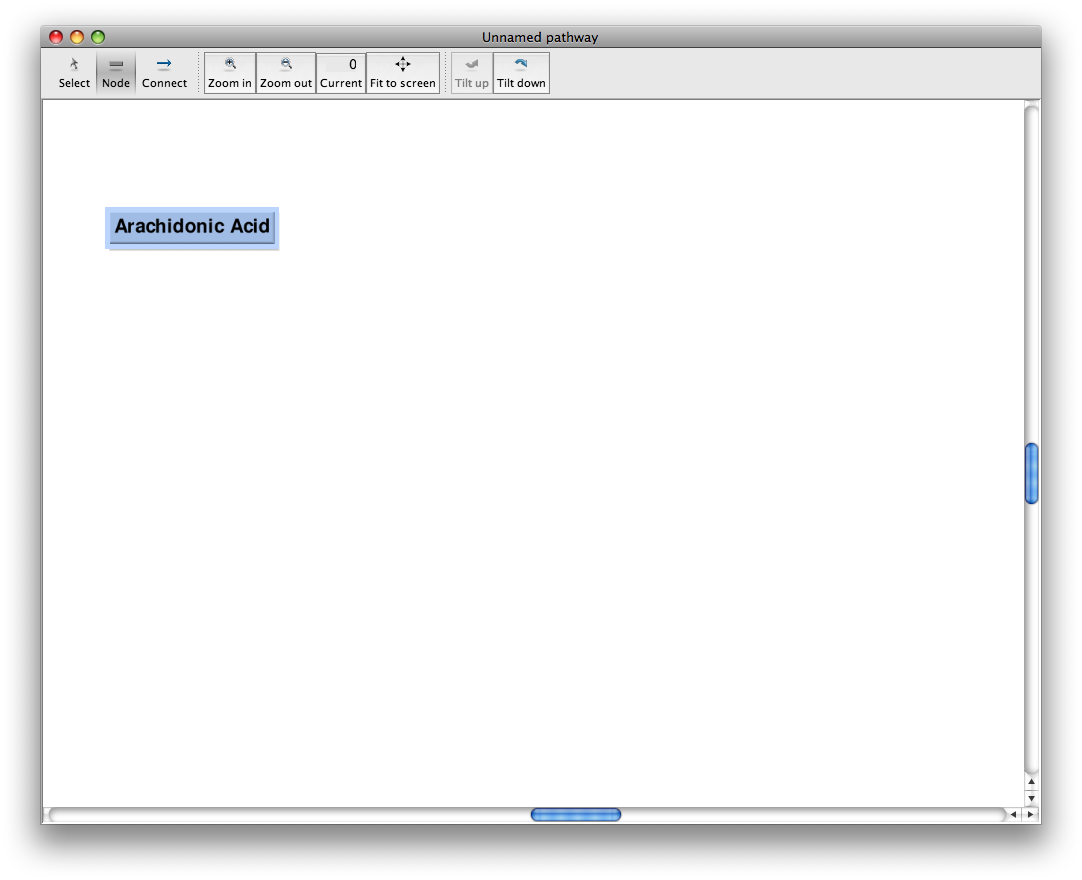
Change the node's label
Change the node's label to anything you want by typing a new value in the "Label:" box in the node's information window. Up to 3 lines may be entered. Additional lines will not be saved. To set line indents, see "Change the appearance of an individual node"

This changes the label on the node's box in the drawing area.
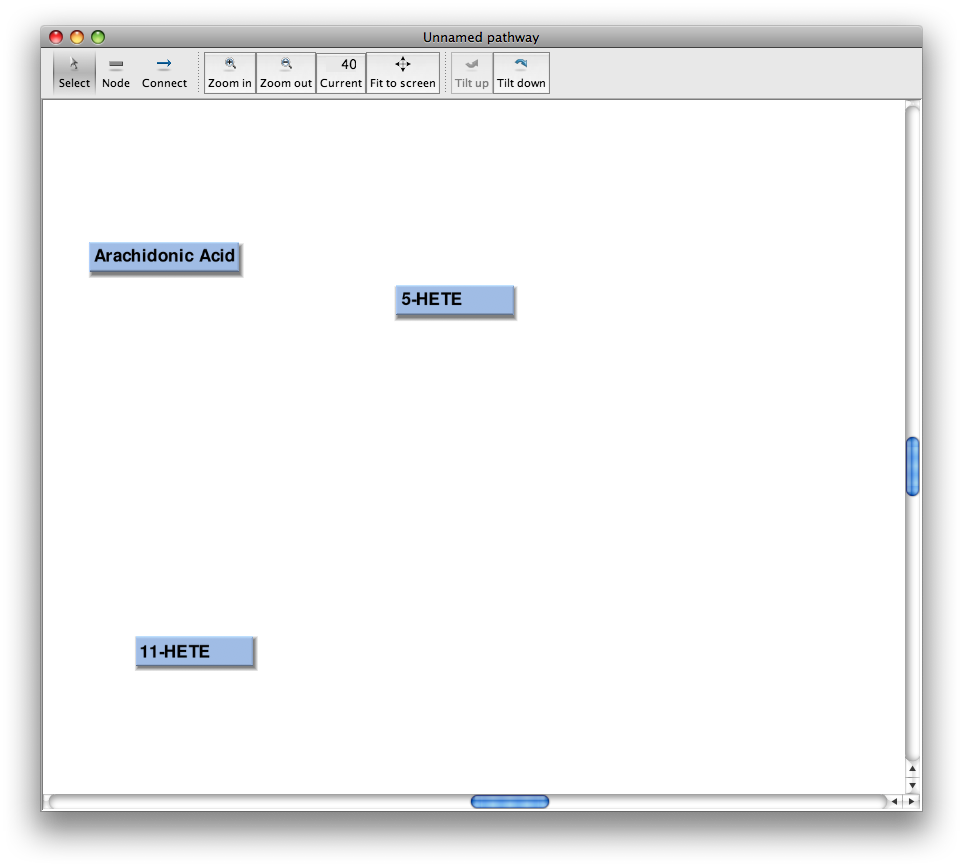
Add more nodes
Continue using the "box" icon. Drop more nodes into the pathway and assign compoundes to them.
Connect nodes to form a process
Click on the dot-and-arrows icon on the toolbar (third from the left) and click-and-drag to draw connections between nodes.
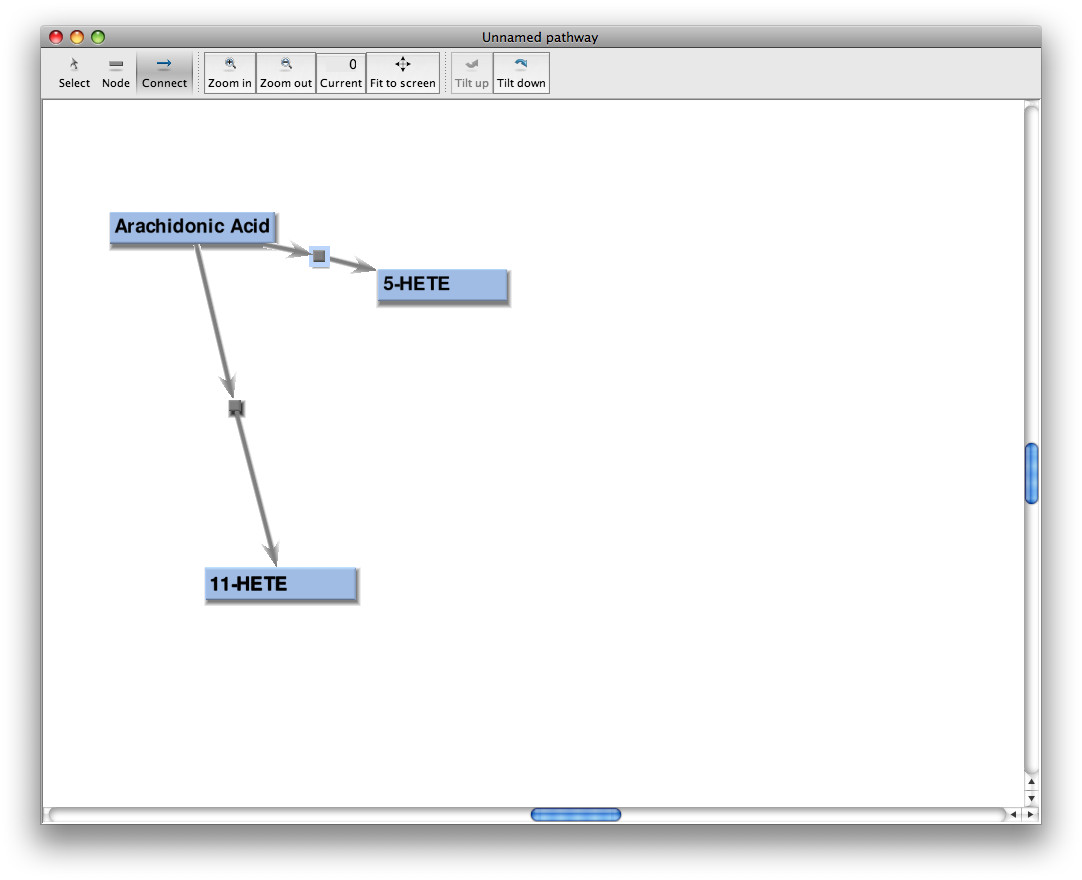
A process or interaction is a connection between two or more nodes. The process is drawn as a square withe arrows connecting the square to nodes. Nodes with arrows into the square are reactants. Nodes with arrows from the square are products.
The first time you connect two nodes, you create the process. After that, you can drag a connection from a node to the dot, or from the dot to a node to add the node to the process.
You can show information about a process by selecting the process square and then performing one of the following:
- Select "Show process info..." from the "Edit" menu.
- Press the right mouse button and select "Show info..." from the context menu that pops up.
- Double-click on the square.
All of these show an information window about the process.

From this window you can enter information on the process in the "Label" field, change its process type, and see a list of the nodes connected to the process. For each of these nodes, you can change their role in the process and their stoichiometry. A node's role can be a reactant, a product, a catalyst, an inhibitor, and so on. Inhibitor nodes are drawn with a "bar" on their connection line. The rest are shown with arrows.
Rearrange nodes and processes
Click on the arrow icon on the toolbar (the left-most button) to enter selection mode. Click and drag to move any node. Process arrows stay attached. Click and drag a process square to move the square.
Shift-click to select multiple nodes. Or click on the background and drag open a selection box around nodes and processes to select a group, then move them as a group.
Change the appearance of an individual node
Right-click on a node. Select "Node preferences..". The "Edit viewing preferences" window appears, with the tab for the type of node selected. The tab contains current viewing preferences for the node. Change any of the settings and press "Apply". Then press "Done" or exit the window.
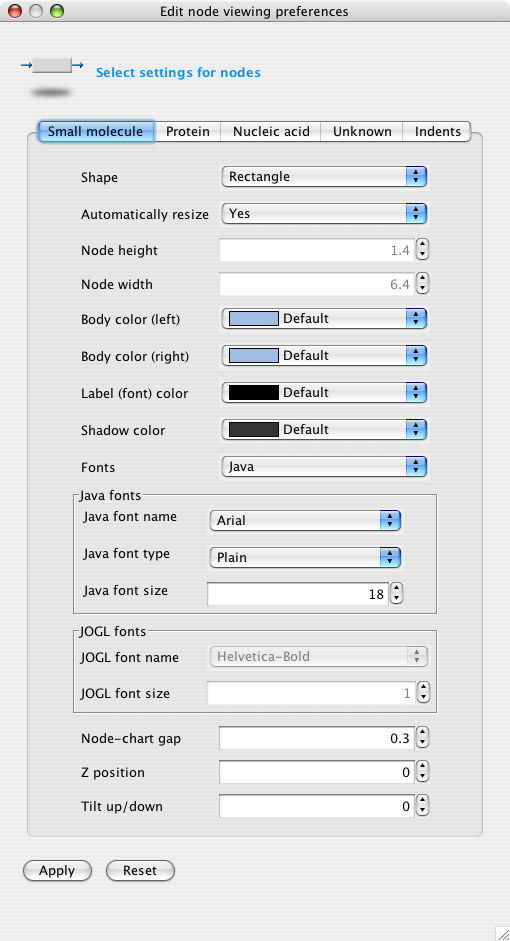
"Automatically resize" means to let the application change node size when typing in a label. If the setting is No, the Node height and Node width may be changed using the drop-down lists. In addition, the handles on the sides of the node are then activated, allowing the user to drag out the sides to a desired position.
The left indent positions of label text may be set using the "Indents" tab.
Change the appearance of all nodes in a pathway or subpathway
Until experience is gained with the Editor, it may be best to limit use of this feature to the very first step in creating a pathway.
Select a region of the drawing area by clicking in the drawing panel and dragging to create a rectangle enclosing the nodes to be changed. Then right-click on any node. The "Edit viewing preferences" window appears. The window contains current viewing preferences for the first of each kind of node in the selection. Changes made to any of the tabs in this window will affect all the nodes in the selection (not just the first node).
Alternatively, select all nodes by accessing the Edit | Node preferences menu. An "Edit viewing preferences" window appears. Changes made on this window will affect all current and new future nodes in the pathway.
Make the desired changes and press "Apply" followed by exiting the window or by clicking "Done". The changes should appear in the drawing panel.
Change the appearance of all processes (interactions) in a pathway or subpathway
Until experience is gained with the Editor, it may be best to limit use of this feature to the very first step in creating a pathway.
Make sure that no processes are selected and go to the Edit menu and select "Interaction preferences...". Alternatively, select a region of the drawing area by clicking in the drawing panel and dragging to create a rectangle enclosing the processes to be changed. Then right-click on any process and select "Interaction preferences". The "InteractionEdit viewing preferences" window appears. The window contains current default viewing preferences for the program, or for the first interaction in the selection. Changes made to any of settings in the window will affect all the interactions in the selection and all interactions that are added later.
Make the desired changes and press "Apply" followed by exiting the window or by clicking "Done". The changes should appear in the drawing panel.
Change the appearance of an individual process (interaction)
Right-click on a process square. Select "Interaction preferences..". The "Edit interaction preferences" window appears. Change any of the settings and press "Apply". Then press "Done"
Change pathway display settings
The Editor uses different sets of settings for displaying pathways, depending upon the type of file. Both sets can be inspected by selecting "Pathway preferences" on the Edit menu.

Changes to pathway settings must be made before reading a file or downloading a pathway.
For .path files, the Editor automatically fits the pathway to the screen dimensions. It automatically uses the zoom setting that was set when the file was saved. Alternatively, the file zoom may be ignored, or a different zoom may be applied, using the Pathway preferences window.
For KEGG file downloads, the same features are available, with the exception of using a file zoom setting. KEGG pathways files do not contain zoom levels.
Node label fonts
By default, node labels are written in Arial 18 point Plain font as contained within the Java language. Font attributes can be changed using the Node preferences dialog.
Labels written in "JOGL fonts", available by way of the Node preferences dialog, appear garbled on some monitors. Java fonts are recommended. However, please be aware that there may be slight differences in the font layout on Windows and Macs.
Manage data display
Construct a pathway and load one or more experiments that contain data to be displayed. Go to the Tools menu and select "Manage data display".
The window that appears contains controls that allow changes to default display settings for data. Make the desired changes on each tab and press "Apply". The changes should appear instantly.

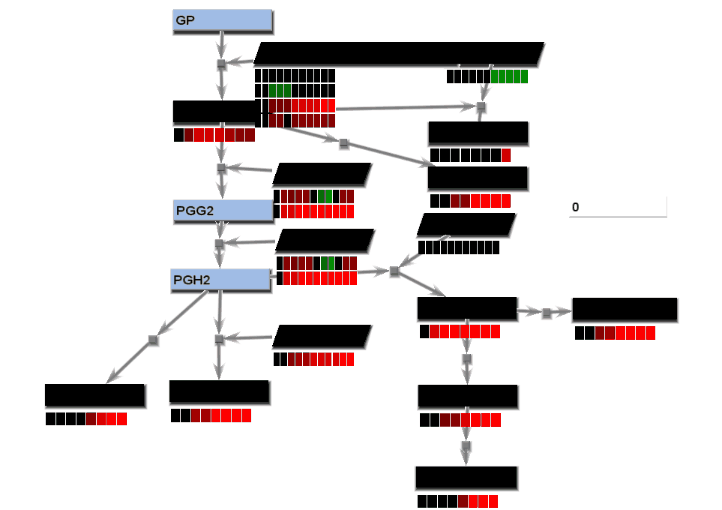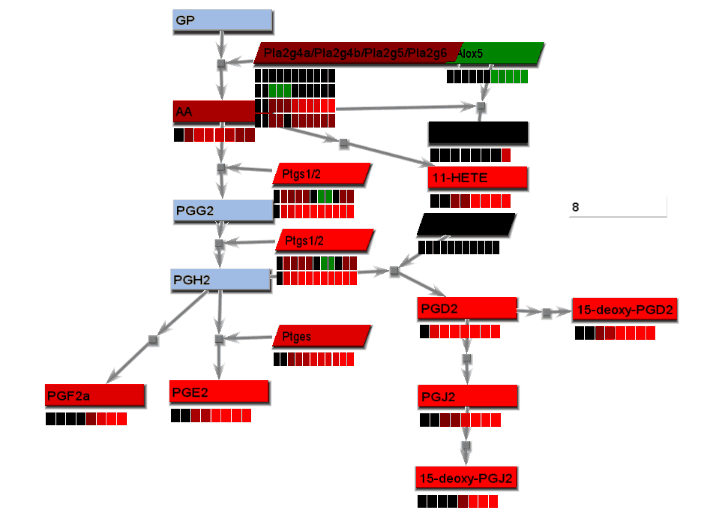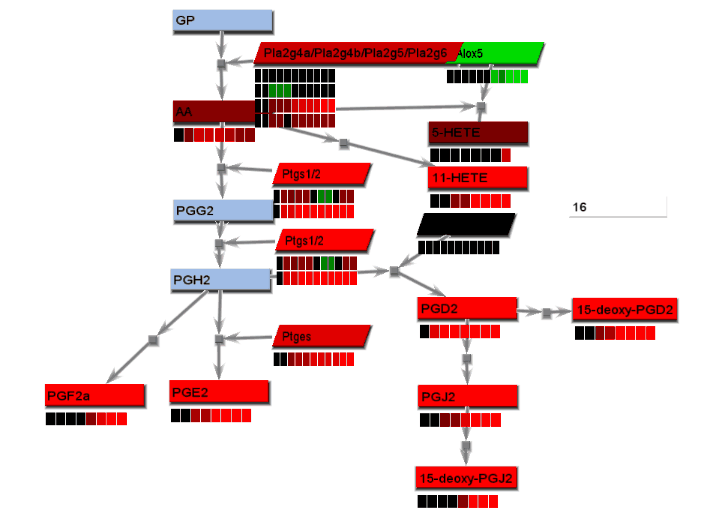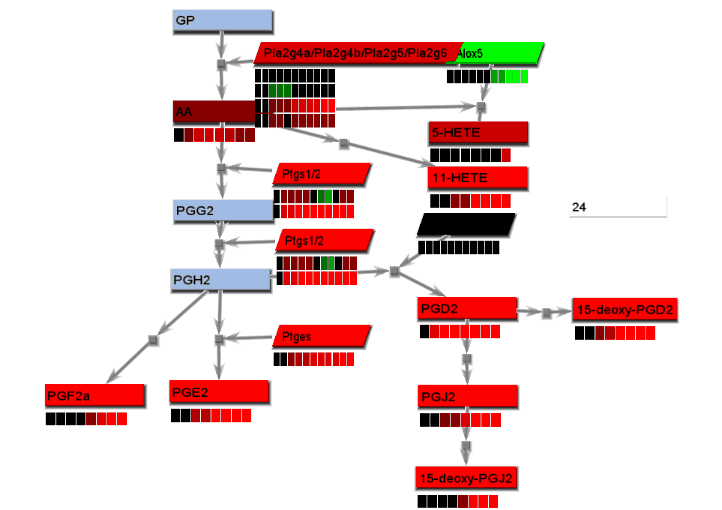
Data may be displayed in the form of heat maps or line charts, separately for each type of node. If heat maps are selected on the first tab of the window, the treatment ratio may then be selected. The default treatment ratio is Kdo/Control.
Alternatively, line charts may be set on the first tab, and line chart settings may be set on the "Chart settings" and "Series colors" tabs.
The changes apply both to data displays in the drawing panel as well as in node information windows.

Obtain an expanded chart for printing and saving to file
On the drawing panel, double-click a chart. In the window that appears, select the "Plot" tab and double-click the chart. Yet another window containing a chart appears, that may be resized. Right-click on the chart and select one of the menu items. "Save as" leads to a window that permits saving a chart in a .png image file. "Print" leads to a window that allows access to the local printing network.


The display properties of the resizable window may be changed using the "Properties" menu item.
Animation
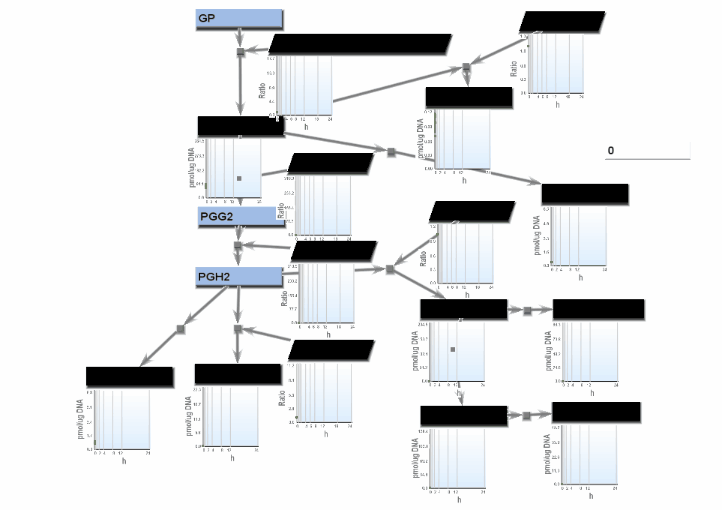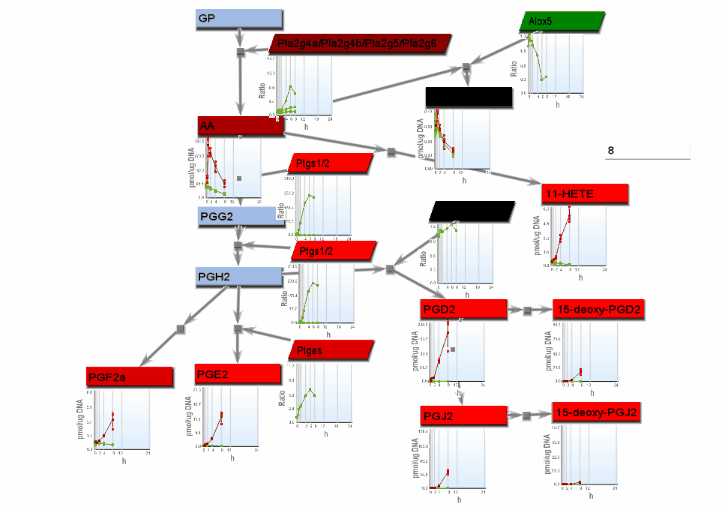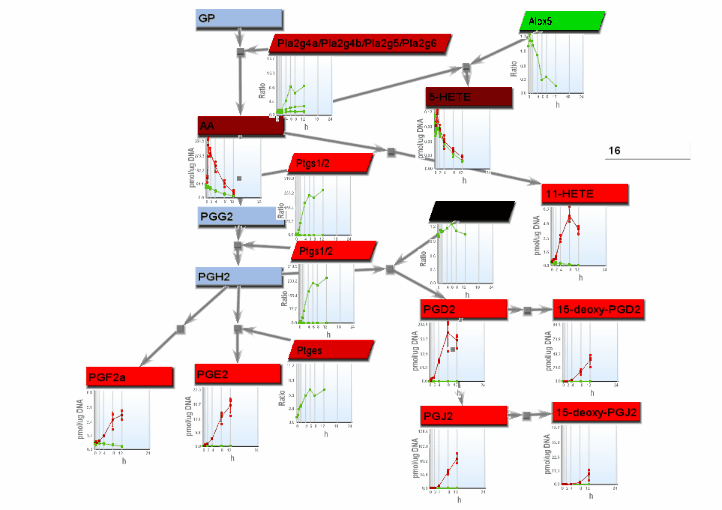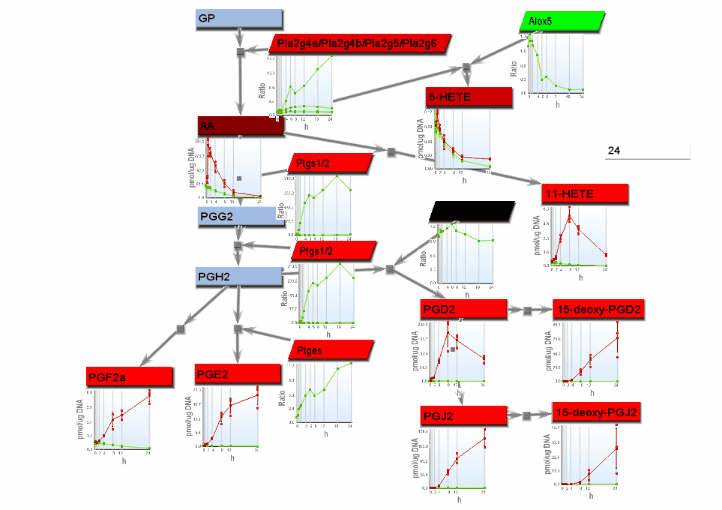
The Pathway Editor provides for animation by changes to node colors, bar charts, heat maps, and xy line charts. Animation is started, stopped, paused, stepped forward, or stepped backward using the toolbar or by selecting options on the View menu. Animation settings are set on the "Manage data display" tool available from the Tools menu. The tabs on this tool that apply to animation are the Basic settings tab and the Animation tab. Animated gifs may be incorporated in PowerPoint presentations.
- The default animation mode is node color changes. Ratios of treatments may be selected for both metabolite data and array experiments. If a single treatment of an individual metabolomic species is selected, ratios with respect to time 0 are animated.
- Bar charts are drawn when "Line charts" buttons are set on the Basic settings tab and the "Heat map/charts" button is set on the Animation tab. This setting may provide the most dramatic visual display of animation.
- Label colors during animation may be set on the Animation tab in order to compensate for a label background that is the same color as the text.
- The total duration of the animation is equal to the product of the time increment (in hours) and the number of times the display refreshes. The default duration is 2 hours times 12 refreshes, or 24 hours.
- If the "Progessive" radio button on the Animation tab is selected, heat maps and xy line charts show progressive changes with time, with rectangles or lines drawn from left to right. Select "Heat maps" or "Line charts" on the Basic settings tab for the desired progressive animation.
Animated gif images
Animated gif images that show changing experimental data are produced using a combination of dialogs. A number of static images, one for each time point, are generated and combined into an animated gif image.

- From the File menu, select "Save image". On the Save image dialog, set the dimensions of the output images. Select the image type. Any type may be selected. However, remember that tif images may be large.
- From the tools menu, select "Manage data display" to bring up the data display settings dialog. There are 4 basic animation modes: node color changes and bar charts, both of which show data at a single changing time point, and progressive heat maps and progressive line charts, which show all data as time progresses. The settings for these modes are:
- Node color changes: Select "Node colors" on the Animation tab.
- Bar charts: Select "Line charts" on the Basic settings tab and "Heat maps/charts" on the Animation tab.
- Progressive heat maps: Select "Heat maps" on the Basic settings tab and "Heat maps/charts" as well as "Progressive" on the Animation tab.
- Progressive line charts: Select "Line charts on the Basic settings tab and "Heat maps/charts" as well as "Progressive" on the Animation tab.
Avi videos
Videos may be created using the "Create avi videos" tool from the Tools menu. This tool operates much the same as the Animated gif dialog. The end result is a file named "anim.avi." The output has been tested using QuickTime and Windows Media Player. Avi videos may be inserted into Power Point presentations as well.
The default settings for a video of 13 frames at 96 dpi image resolution generate a file that is 27 Mb in size. A very high video resolution is possible by selecting tif images and setting the resolution to a large dpi, such as 300. However, considerable RAM is necessary for processing. In addition, the video size may be on the order of several hundred megabytes!
Sample .avi videos: nodecolors.avi (27MB) progressiveheatmaps.avi (27MB) progressivelinecharts.avi (27MB)
- Read data from files or download data from a database.
- Construct a pathway, or open a pathway file.
- From the Tools menu, select "Create animated gif image" to bring up the Animated gif dialog.
- Set the output directory using the "Set directory" button.
- Set other values of the time course: start and stop times, the time increment for the data, the number of steps, the time between frames as they will appear in the gif, and whether or not the image will repeat.
- The time between image acquisitions is set at 3 seconds to allow files to be written between refreshes of the panel. This may be increased if it is discovered that some time points are not shown in the gif.
- If the user wishes to keep the intermediate files obtained at each time point, set "Remove temporary files" to No.
- A node showing the time may be created using the "Add time node" button. The time node may be moved and other properties changed on the panel.
- Press "Start" to start the process. If the user wishes to interrupt the process, press "Stop."
- Note that not all of the changes on the viewing panel and tool bar will appear in the resulting gif.
- A dialog will appear at the end of the process, notifying the user that an image file named "anim.gif" been generated in the output directory.
Save a pathway as a *.path file
Go to the "File" menu and select "Save pathway as..." to get a save window. Enter a file name, select the "PATH" file type and click "Save." The PATH format is our own file format that supports saving display parameters for the pathway and its assigned compounds.
Experiments are also referenced in the .path file if experiments were previously downloaded from the database. The data for these referenced analyte (lipidomic), microarray, and protein array experiments are automatically downloaded from the LIPID MAPS database when the file is read back into the program.
Save a pathway as an image
Go to the "File" menu and select "Save image as..." and select any of 'bmp', 'jpg', 'jpeg', 'png'. Please note that on the Mac, before saving your pathway as an image, you must first select any node in the pathway. If you do not first select a node, the image will be saved, but if experimental data is shown in the pathway, some of this data may be missing from the saved image.
Search for experimental lipidomics data
Go to the "Tools" menu and select "Manage time course experiments..." to get a list of loaded experimental data (initially empty).

Click on the "Add" button to add an experiment from the database.

In the window that opens, optionally enter a keyword or keyword fragment and press the "Search" button to search descriptions and lab titles in the database.

Select an experiment from the list and press the "Add" button to load it into the program. Relevant experimental data is automatically shown in heat maps or line charts under each node in the pathway.
To view an experiment's data, select the experiment and press the "Show info..." button.

Select any of the compounds in the Manage time course experiments window to show their heat map or line chart and raw data to the right.
You can add more experiments to the Manage time course experiments window by clicking the "Add" button and searching the database again. Click the "Remove" button to remove an experiment. Click on the checkbox beside an experiment to show it in the pathway (checked) or hide it (unchecked).
Check the "Average experiments" checkbox to average the data in the list of experiments for purposes of display.
If the "Average experiments" checkbox is not checked, then only the data of the first checked experiment in the list is displayed. The rest of the experiments are ignored.
Search for microarray or protein array experiment data
Go to the "Tools" menu and select "Manage microarray experiments..." or "Manage protein array experiments..." to get a list of experimental data.
The next steps are nearly identical to those in "Search for experimental lipidomics data". The exception is that data is downloaded only for these gene symbols that are currently assigned to existing nodes.
The database is checked for data assigned to gene symbols when an array experiment is selected and the "Refresh" button in the "Manage microarray experiments" or "Manage protein array experiments" windows is pressed, and also when a .path file referencing an array experiment is opened.
P-values for a microarray experiment may be searched using the window that appears when a microarray experiment is selected and the "P-values" button is pressed.
To search P-values, select a type of P-value and enter the upper P-value of the range to be used.Press "Search". A list of gene symbols and their P-values are shown.
The gene symbols and P-values can be sorted by using the "Sort" buttons, or by pressing a column header.
Using Pathway Editor to view user-defined data
The Pathway Editor loads CSV (Comma Separated Value) data files generated by Microsoft Excel. CSV files may be opened using Excel for editing, if desired. Data files may also be created using a text editor. Below is a description of a CSV data file.
Using CSV data files
Note that steps 1 and 2 can be performed any number of times and in any order.
- Go to File | Open CSV data file and load a file ending with the extension .csv. When loaded in Pathway Editor, the data can be viewed using the menu items in the Tools menu.
- Go to File | Open Workbench pathway file and load a .path file. The nodes in the file may have database compounds assigned, or not, as desired.
- The data will be displayed in the Pathway Editor.
- When generating a new node, the Editor will automatically assign data to nodes: First, create a node and assign a node type (Small molecule, nucleic acid, or protein) using the Node information window. Next, assign a label name that is a compound name from the data file. On a Windows desktop, press the "Done" button. On a Mac, close the dialog. If the information has been typed correctly, the data will automatically be associated with the node and displayed.
Rules for writing CSV data files
Each data file may contain one or more experiments. Each experiment may contain Analytes (metabolites, comprised of Small molecules, Proteins, or Nucleic acids), Microarray, or Proteinarray compound types. However, an experiment may not contain a mixture of compound types.
- Start the file with a line beginning "Experiment list".
- The next line should contain at least one leading comma.
- Lines 3 through 10 describe an experiment and should contain:
- A line beginning with the word Experiment.
- A line beginning with the word Date, a comma, and the date in month-day-year format (mm-dd-yyyy). The date in this format must also be surrounded by quotes (that is, July 12, 2008 may be written "07-12-2008"). The date may also be written as mm/dd/yyyy, without quotes (written as 07/12/2008).
- A line beginning with the word Type, a comma, and the type of experiment (Analyte for a metabolite, Microarray for a microarray experiment, or Proteinarray for a protein array experiment).
- A line beginning with the word Ratios, a comma, and a list of treatment type ratios to be used by the program. The ratios will appear on the Manage data display dialog.
- A line beginning with the word Description, a comma, and a brief description of the experiment.
- A line beginning with the phrase Time units, followed by the time units for the experiment (h for hours, etc. These must be the same for all compounds.)
- A line beginning with the word Lab, followed by a description of the lab.
- A line beginning with word System, followed by a description of the biological system.
- Line 11 should contain a leading comma.
- The next 7 lines should contain the description of the compound. The compound should be of the same type as a node in the pathway to which the data is to be assigned.
- A line beginning with the word Label, a comma, and the compound name.
- A line beginning with the word Type, a comma, and Small molecule, Protein, or Nucleic acid.
- A line beginning with the phrase Gene symbol and a comma. If the compound is a protein, the text symbol for the microarray or protein array spot should be next. Leave the symbol empty for other compound types.
- A line beginning with the phrase Data units, followed by the units for the data. This should be the same for all compounds within the experiment.
- A line beginning with the word Treatment, a comma, and the type of treatment. Treament types may be the same as listed in the LIPID MAPS pathway database. Alternatively, a different treatment type may be used on this line, as desired.
- A line beginning with the word Times, a comma, and all the measurement times for the compound, separated by commas. It is not necessary to enter values for time 0.
- A line beginning with the word Measurements, a comma, and all the measurement values for the compound, separated by commas. It is not necessary to enter values for time 0.
- Each compound description should be separated by a line beginning a leading comma.
- Additional guides:
- If the compound is a Small molecule, Protein, or Nucleic acid analyte, the label name in the CSV file must be the same as the label name for the node in the pathway diagram.
- If the compound is part of an array experiment, the compound in the CSV file must have a label name that is the same as the label name for the node. If the node has one or more gene symbols assigned, the compound in a the CSV file must possess one of the gene symbols. If the node does not have a gene symbol assigned, the gene symbol of the compound can be any desired text. In either case, data will be automatically associated with the node.
- Repeat 4 and 5 for each compound in the .csv file.
Simplified user data files
The Pathway Editor reads simplified user data files in Excel and in tab-delimited formats. Both metabolomic (analyte) and array data may be contained in these files. As with CSV files, they may also contain biological treatments that are different from those contained in the Pathway database.
Excel and tab-delimited files are read by selecting the appropriate menu items on the File menu. Upon reading a user data file, the Pathway Editor creates an experiment and assigns data to nodes that contain the label that is the same as a name in the files. If no compound is associated with the node, a new compound is created. This new compound is cleared from the program when the experiment is removed.
When calculating averages for heat maps, and for ratios calculated for line charts, each experiment is weighted equally, regardless of the number of repetitions within the experiment.
Heatmaps involve ratios of measured values at each time point. If there is no measured value for either numerator or denominator, a blank is drawn. Similarly, node color animations also involve ratios of measured values. If there is no measured value at a time point for either numerator or denominator, the last color for which there is both numerator and denominator is used.
Excel data files (Microsoft Excel 97-2003 .xls files)
The format of VANTED-style template 1 (for analyte) and template 2 Excel files (for arrays) is followed (
Example Pathway Editor template files are AnalyteTemplate.xls and ArrayTemplate.xls.
For both analyte and array files, replicate numbers are ignored by the Pathway Editor. In addition, time values need not be integers. Any time unit may be used. However, the user is cautioned when attempting to simultaneously view database and user data, as the time unit of the first experiment in each experiment list is used for charts and heat maps and may not be the same as for other experiments.
Excel analyte files
Some optional modifications of the template 1 format have been added for the Pathway Editor, and should be readily understandable by inspection. These include differences in required and optional fields. Optional fields are marked by an asterisk (*).
- In VANTED, template 1 files (for analytes) are centered around species and genotypes, while the Pathway Editor is more concerned with perturbation experiments, in which a single cell or organism is subjected to one or more treatments. Template 1 files also differ in that the type of analyte (Small molecule, Protein, or Nucleic acid) may be entered in the analyte type cell (B9). If this cell is empty, analytes are assigned a default cell type of Small molecule.
- The format of Excel dates used by VANTED is different from the format used by Pathway Editor(VANTED: dd/mm/yyyy vs. Pathway Editor: mm/dd/yyyy).
Excel array data
A description of the Excel data format for microarrays and protein arrays may be found at http://vanted.ipk-gatersleben.de/index.php?file=doc9.html (VANTED "Template 2"). This format follows that of the program J-Express. Pathway Editor will assign the data to protein nodes.
The initial rows of the file may contain information that is ignored until a row beginning with "Spot" or"spot" is reached. This row is assumed to contain column headers. Column headers are traversed until data column headers are found. The columns prior to reaching the data columns are assumed to contain metadata for the protein. Cells underneath data column headers contain data for the time and treatment specified by the respective column header.
Data column headers may contain the following different formats:
- "nncccc_nn" where "nn" represents a time that may contain a decimal point. "cccc" is a string of one or more alphabetic characters. These characters may be a single treatment, or it may be a ratio of 2 treatments separated by a forward slash (for example, Treatment/Control). "_" represents an underscore. The last characters represent a replicate number that is ignored by the Pathway Editor. Representative headers are "0Treated_1" and "1.0Control_2".
- "nn cccc" where "nn" represents a time that may contain a decimal point. A blank space follows. "ccccc" represents a single treatment, or it may represent a ratio of 2 treatments. Representative headers are "0 Treated/Control" or "1.0 Treated/Control". This is the format that used in the example file ArrayTemplate.xls.
- "nncccc" where "nn" represents a time that may contain a decimal point. "cccc" represents a single treatment, or it may represent a ratio of 2 treatments. Representative headers are "0Treated/Control" or "1.0Treated/Control.
When an Excel array data file is read, a dialog appears that displays text fields in which experiment metadata may be entered. An important concern is that the column to be used for comparing with node labels must be carefully chosen. In addition, if the column headers contain single treatments, and one or more ratios should be chosen for display. The Pathway Editor computes ratios as needed. If data column headers contain treatment ratios, the selection list is non-editable, and all treatment ratios in the list are accessible by the user.
Metadata for the respective protein is shown on the Comments tab of the compound dialog for each node.
Tab-separated values (.tsv) files
- Tab-delimited analyte data files
Tab-delimited analyte data files contain 4 columns with the following headers in the first row: "Analyte", "Treatment name", "Time", and "Value". Each of the columns must be separated by tabs. Times and values may be in either integer or decimal format. An example is contained in SampleTabDelimitedAnalyteData.tsv. This file contains 4 treatments. The treatments may be any desired by the user.
When a tab-delimited data file is read, a Metabolomics experiment metadata dialog appears in which the user must select or enter the date of the experiment, the analyte type, time units, data units, and one or more ratios for heat map or chart ratio display.
- Tab-delimited array data files
An example file containing array tab-delimited data is SampleTabDelimitedArrayData.tsv. This file contains 10,000 rows of data drawn from another file containing 43,962 lines of data for an experiment involving RAW cells. Additional information on this experiment may be found at http://www.lipidmaps.org/data/results/raw2647/kdo2lipidatimecourse/compactin.html. Attempting to read a file of this size may cause problems. If this happens, reducing the number of lines in the file may be effective.
The first row contains column headers. A varying number of metadata columns may be contained in array data files; however, one of these columns must be headed "Gene symbol" and must contain names of node labels if the data is to be associated with a node. To the right of the metadata columns are the actual data columns.
The format of the data columns is "nn cccc", where "nn" is a time and "cccc" is a single treatment or a ratio of treatments (such as "Treated/Control"), with time and treatment separated by a space. A zero time column is not necessary, though it may be included.
After a tab-delimited array data file is read, an Array experiment metadata dialog is shown. The user must select the array type (Microarray or Protein array) and enter text for the time units. If the file contains single treatments, ratios must be selected from the list of ratios.
Metadata for the gene symbol is shown on the Comments tab of the compound dialog for each node.
Pathway Enrichment Analysis
The Pathway Editor can analyze a list of LIPID MAPS identifiers and report on the involvement of KEGG pathways using a technique known as "pathway enrichment analysis".
The pathway enrichment analysis tool is accessed from the Tools menu, as follows.
- Paste a column of LIPID MAPS lipid identifiers into the text area of the tool.
- Press "Analyze" to send the IDs to the Pathway database, where a function determines the presence of these identifiers in KEGG metabolic pathways. There are 145 metabolic pathways in the Pathway database (as of November 14, 2012).
- The results are sent back to the program, which calculates the probability of enrichment of each pathway using the hypergeometric distribution (A. Hsaio et al., Nucleic. Acids Res 2005, vol 33, W627-W632) and displays the results in the tool. A smaller p is an index of the reliability of the calculation (S. Gupta, personal communication).
Following are the parameters used to calculate probabilities, using one type of notation:
- N is the total number of LIPID MAPS identifiers in all KEGG pathways.
- k is the number of LIPID MAPS identifiers submitted (in the examples below, k is 30).
- b is the number of LIPID MAPS identifiers existing within a particular pathway.
- i is the number of LIPID MAPS identifiers within a particular pathway that are contained in the submitted list.
Results are reported in a format in which N is on the first line and k is shown on the second line. Below these are the results for each pathway, in decreasing order of i. Each line contains the name and KEGG ID of the pathway, i, b, and the p value as a decimal value between 0 and 1.
Following are 2 test sets of 30 LM identifiers. The first set was chosen randomly. The second set was chosen from the set of identifiers that are contained within the KEGG arachidonic acid pathway.
Graph Modeling Language (GML) pathway layout files (.gml)
The Pathway Editor reads and writes pathway layouts in GML format. These layouts describe a two dimensional node layout, along with node labels. VANTED and other modeling programs may use more features of GML, but such information does not interconvert into Pathway Editor-usable information.
Two GML files are available: CentralMetabolismBarley.gml and VANTEDDemo.gml. Both were obtained from the VANTED program (http://vanted.ipk-gatersleben.de/index.php?file=doc0.html).
A GML file may be read after selecting the appropriate item from the File Menu to bring up a file dialog. The Pathway Editor will read the file and present a "GML reader dialog". By default, nodes are assumed to be of small molecule type. The GML reader permits the user to change the type of selected nodes as desired. Pressing the "Layout" button will transfer the layout to the drawing panel and will connect nodes using edge directions that are specified as reversible or non-reversible in the file. The layout will be expanded or compressed to reside within the borders of the viewing panel, unless preferences are set differently on the Pathway preferences dialog available from the Edit menu.
A GML file may be written by selecting the appropriate item from the File Menu to bring up a file save dialog. The Pathway Editor will write edge directions and relative node layout in the output file in a way such that it will be readily laid out and displayed upon reading by VANTED. VANTED deals with interaction glyphs differently than the Pathway Editor. An interaction that has more than 1 reactant or more than 1 product in a GML file generated by the Pathway Editor does not display properly in VANTED. Node colors will also be incorporated into the file. However, references to subpathway nodes or nodes contained within subpathways are not included in the GML file.
Drawing organelles
Organelle drawing makes use of complex 3D drawing and lighting effects.
- Select the "Organelle" tool button, and click anywhere on the drawing panel. Press the "Select" button to stop creating organelles.
- When created using default settings, organelles are divided equally in the + Z and - Z directions, that is, at Z = 0, with half the organelle above the XY plane and half above. The XY plane is the surface of the monitor. The default mode setting is "Points." Other settings are "Filled" and "Filled no shading." Shapes include ellipsoids and spheres, cubes and rectangles, and cylinders.
- Change viewing options in the same manner as for nodes and interactions: On the "Edit" menu, select "Organelle preferences" to set features of all organelles, or right-click on an organelle and select "Organelle preferences" to obtain a dialog enabling changing features of a single organelle.
- Nodes are created by default at Z = 0, so they are easily placed within the organelle volume. They may be viewed through the "Points" and "Filled" modes. In "Filled no shading" mode, it may be necessary to decrease the organelle opacity (for example, to 20) in order to view nodes.
- To place a pathway within a drawn organelle, one may create the pathway and organelle separately on the drawing surface, and then move one to overlay the other. Alternatively, one may make the organelle invisible using a setting on the View menu, draw or modify a pathway, and then make the organelle visible.
- To move an organelle with a pathway contained within it, outline the organelle using the mouse to select the organelle and the pathway's nodes and interactions. Then place the mouse on the organelle, press the left mouse button, drag the selections to the desired screen location, and release.
- The XY dimensions of organelles can be changed with handles, similar to changing height and width of nodes. The Z setting changes the depth of the node.
- Use the "Tilt up/down", "Rotate around Z", and "Tilt left/right" preferences to orient the organelle in 3D space.
3D Drawing
3D drawing
When reading a pathway containing organelles, nodes, and interactions at values of Z greater than 0, it may be necessary to deselect the "Automatically fit to screen" option on the Pathway preferences dialog. This feature cannot effectively fit structures outside the XY plane at this time.
Using compressed pathways and subpathways
1. A compressed pathway may be created by selecting a group of participant and/or subpathway nodes, right-clicking on the drawing surface, and selecting "Compress selection".
The selection must include at least 2 nodes and at least 1 interaction.
The resulting subpathway node has display properties that may be set by right-clicking and selecting "Show info" or "Subpathway preferences", analogous to participant nodes.
Interactions may be drawn to and from subpathway nodes using the mouse if they exist within the same upper-level pathway and are showing. "Links" or individual connections may be made between participant nodes or subpathway nodes in different pathways using dialogs. Connections may also be made between participant and subpathway nodes node in one pathway and an interaction in another. These links are either created anew, or are added onto already existing interactions.
When an interaction is created between a node in one pathway and an interaction in another pathway, the resulting interaction is considered to be within the same pathway as the original interaction.
2. The subpathway node contains a pathway that can be expanded by right-clicking, and selecting "Expand selection". Note that this feature can result in loss of information pertaining to the sub pathway. Expanding subpathways using the Manage subpathways dialog should be the preferred method.
3. A second way of expanding can be executed from the Manage subpathways dialog, obtained using the Tools menu. This dialog displays tabs that contain the following:
- A list of all subpathways contained within the top level pathway.
- For the top pathway and for each subpathway, a panel showing the nodes contained within each.
To expand a subpathway, select a subpathway on the pathway list tab and press "Expand selection."
To return to the top-pathway, press "Show top pathway".
Information on the subpathway can be obtained by selecting a subpathway on the pathway list tab and pressing "Show info."
4. The contents of a subpathway can be altered by bringing up the subpathway on the display using one of the above expansion methods, and altering or changing the display.
The changes are stored when the user displays another subpathway, or the top pathway.
5. Links or interactions between nodes and interactions that are internal to subpathways may be viewed and deleted using the "Manage pathway links dialog", available from the Tools menu.
To view properties of a link, select a row of the Manage pathway links table and press the appropriate button. To delete a link, select a row and press "Delete link". There is a bug in the program involving a link between a node in one pathway and an interaction in a second. In this case, select the row again and press "Delete link" a second time.
Subpathway interactions
Interactions may be constructed from any participant or subpathway to any other participant or subpathway. Reactants and products can also be added to an existing interaction between or within subpathways.
- The user can draw connections between drawn subpathway and participant nodes and interactions using the mouse.
- To construct interactions between components that are not currently displayed, go to the "Links to outside" tab of the subpathway information dialog. This tab shows current interactions between internal and external nodes for the subpathway.
- Press "Add interaction." The Create link dialog is displayed.
- The Create link dialog contains 3 lists.
- The first list contains selections for the target subpathway that contains the product of the link. "None" means that the product components that are shown in the third list at the same level of compression as the source pathway.
- The second list contains all the options for the component that may comprise the reactant (or an interaction at one end) in the link. These are within the source pathway. Interactions may not connect directly to interactions.
- The third list contains all the selections for the component that may comprise the product (or an interaction at the other end) in the link. These are within the target pathway that is selected in step 1.
- Select the desired component from each list and press "Create".
- The interaction will be created.
- Note that when the target component is not showing on the display, the interaction may not show on the panel. This is because drawn interactions may be misleading as to the actual interaction participants at different levels.
Check dialogs to ensure that the interaction has been been created. Interactions created using the Create link dialog show only when the internal pathway components are in the first internal pathway of the subpathway node. - Lists of participants on the Interaction dialog may appear differently depending upon whether they are created in steps a to e, or by compressing an area of the drawing panel.
- The properties of the interaction can be changed on the dialog for the interaction, obtained by selecting the link on the "Links to outside" tab and pressing "Show interaction". The link can be deleted by pressing "Delete link".
3. After a subpathway is expanded on the drawing surface, changes may be made to the subpathway. These changes are preserved after the modified subpathway is compressed (i.e., another pathway is expanded). Information on any visible dialogs is then updated.
How to construct a layered pathway:
Two methods may be used. The first is laborious and involves adding nodes and interactions individually. Z heights that place icons at different heights above the plane of the screen are entered during construction. The second method is faster and involves prior construction of pathways for the layers and saving to separate files, depending upon the desired result. After the files have been constructed and saved, build up a layered pathway as follows:
- Depending on the result desired, you may wish to unselect "Automatically fit to screen" on the Edit | Pathway preferences dialog
- Go to View | Manage layers and open the Manage layers dialog.
- On the Manage layers dialog, press "Add layer" and open a file containing the first layer. This becomes layer 1 in the table.
- The contents of each subsequent file that is opened gets added as the next layer.
- The same file can be used for different layers.
- Select layers in the table and move the slider to the desired Z height for each. Use the "Unselect" button to unselect the layer.
- Use the Tilt up/Tilt down buttons and the vertical and horizontal slider controls on the main viewing panel to position the whole pathway.
- To change viewing preferences of entire layers, select the layer in the table. Use the "Set invisible" button to make the selected layer invisible or visible. Move the selected layer around the viewing panel with the mouse. Right-click a component of the selected layer in the viewing panel and modify the viewing preferences of this component type in the layer as desired.
- To change the properties of an individual component within a layer, select the component. Move the component in the XY plane with the mouse. Right-click on the component and modify viewing preferences.
- When the layered pathway is saved, layers are also saved.
- To clear the dialog, go to the File menu and select "New pathway."
Opening a layered pathway:
- To open a layered pathway, go to Edit | Pathway preferences and unselect "Automatically fit to screen" on the Manage pathway preferences dialog.
- Open the file containing a layered pathway as you would normally open a pathway file.
- The Manage layers dialog can be used to add additional layers to a layered pathway opened in this manner.
SBML Support
Constructing, reading, and writing models in the Systems Biology Markup Language (SBML)
SBML functions
The Pathway Editor can read and write models in Systems Biology Markup Language Format (http://www.sbml.org). SBML models are quantitative computation models that are mathematical descriptions of time-dependent changes to species in a pathway. The Pathway Editor makes use of JSBML, a Java-based API that is still under development (http://sbml.org/Software/JSBML). The complex nature of such models requires that users possess a good understanding of SBML before modifying or building models. Seven different releases of SBML specifications are supported. Each has a level number and version number. Level 1, version 1 models are read-only; new models in level 1, version 1 cannot be written. To ease use, dialogs in the Pathway Editor may appear different for different releases. In addition, some menu items and controls are inactivated (greyed out) to indicate that they do not apply to the release that has been selected. In the following, note that information entered into dialogs is inserted into an SBML model when the user presses "Done" on Windows or closes a dialog on Macs.
Reading (importing) an SBML model
- To read a model, go to File | Open SBML model. Participants in SBML are referred to as species, while the Pathway Editor contains 4 kinds of participants. There are 4 menu options for creation of pathway participants from SBML species in a model file, depending upon the user's choice of how to handle SBML species modifiers: as proteins, small molecules, nucleic acids, or unknown. For biologists, proteins are a very likely option for species modifiers. Using the Open file dialog, navigate to, and select, a file with the extension .sbml or .xml.
- The model in the file will be read by the program. SBML species characteristics will be displayed in an SBML Reader dialog window. The user can check a row, and, using the mouse, left-click on the view panel and selecting Edit | Paste to create a node on the panel that contains the species at the location of the mouse click. Alternatively, pressing "Center node" creates the node at the center of the view panel. The row on the table will then become non-selectable. The newly created node can be moved to a subsequent different location using the mouse. Interactions with correct connectivity will be added automatically, once nodes for the interaction have been created.
- The user may use the SBML Reader to:
- Set the last cell in each row to a different default participant type if the species is not used as a species modifier. The default type may be changed using the list below the table. Alternatively, the user can set the desired type of participant on the last cell of each row of the table before selecting the species to be pasted.
- Set the information to be displayed on the node label: species ID, species name, or species type.
- Clear the drawing panel by pressing "Reset" to start over.
To reiterate, the user can arrange nodes in the drawing panel by recursively selecting (checking) the left cell in a row and left-clicking on the drawing panel and pasting by going to Edit | Paste on the top menu to achieve the desired layout. Alternatively, the user can press "Auto layout" to lay out nodes automatically. The viewing panel can be cleared by pressing "Reset" on the SBML Reader dialog.
- If the model has been created using the Pathway Editor, pathway layout information contained within the model will be used to draw the pathway. However, species that have been added outside the Pathway Editor will not be shown, and it will be up to the user to create nodes individually. Pathway participants that do not have species associated with them will not be shown in the SBML reader dialog.
- After adding all species to the pathway drawing panel, the user can press "Update compounds" to update compound information from the LIPID MAPS Pathway database.
- At any point before or after adding nodes to the drawing panel, SBML model information may be viewed using menu items available from the SBML menu. In particular, the Tools menu has "Manage nodes" and "Manage processes" options that cause list dialogs to be displayed. These dialogs show the relationships between LIPID MAPS pathway participants and interactions, and SBML model entities that have been assigned.
- Nodes in each SBML compartment can be colored similarly if desired. On the File menu, select "Compartments | SBML" to create a dialog that achieves this. To view compartments and compartment types on the drawing panel, select "Compartments | SBML" on the View menu.
- If an SBML reaction in a file being imported has no product species reference, the Pathway Editor draws an interaction glyph with reactants, but there is no product node. This situation occurs, for example, when a species is degrading to an undefined product species that is not a part of the list of species in the file. The interaction glyph in this case is drawn only when the automatic layout feature is used.
Creating or modifying SBML model information
- An SBML model may be created after Pathway Editor nodes and interactions are added to the viewing panel. The Model description dialog must be the first dialog that is accessed. The other dialogs on the SBML menu are not available until this is done. Fields marked with an asterisk are required. All others are optional. On the Model information dialog, select the level and version of the SBML model to be created, and enter a name for the model, such as "Eicosanoid metabolism". Press "Create". If creation is successful, a success message is presented, and greyed-out sections of the dialog then become accessible. The Model information dialog allows entering information on creation date and model creators. These are components of the model history.
- Model history in level 3: When a model is created, the time and date is shown on the Creator panel of the Model information dialog. The time is Universal time (GMT). The time can be shown in local time by press "Show local time". After a model has been saved to a file, the model adds a modification date and time whenever the "Done" button on the dialog is pressed. This cannot be altered. However, to prevent needless accumulation of dates, dates can be deleted from the table on the "Modification dates" panel. The user can select any of the rows and press "Remove selected". The modified file can then be saved. The latest date can be seen when the user next reads the file.
- The third tab on the model information dialog shows unit definitions that are preset in SBML. The user can set default unit definitions by entering newly constructed unit definitions IDs into the text fields. A species conversion factor can also be set. Buttons on this tab bring up the UnitDefinition dialog that can be used to set default units. Alternatively, the ID of previously created unit definitions can be typed into text areas. The conversion factor allows setting a parameter that allows calculating the effect of a reaction on a species. The parameter is set by way of a drop-down list.
- SBML species and reactions are created by accessing and selecting from list dialogs, or by using dialogs obtained by right-clicking pathway nodes and processes (interactions). Note that once an initial SBML species or reaction has been created or deleted, the SBML reader dialog is cleared.
- To add a species, first add one or more Compartments. Next, create a node on the drawing panel. Then, right-click on the node and select "SBML species info". Alternatively, select the node on the SBML | List of species dialog. The Species dialog for the node will be shown. The user can enter (or update) information. Pressing "Done" will save the data.
- To add a reaction, first ensure that all the participant nodes in a reaction have SBML species associated with them. Then, right-click on the reaction glyph and select "SBML reaction info". In the Reaction dialog, ensure that the desired roles for the species are entered on the "Species references" tab, and press "Assign species references". Press "Species reference details", enter information concerning species references on the resulting dialog, and press "Done". Finally, press "Done" on the Reaction dialog to save the information.
- Most dialogs for entry of SBML information allow entry of MIRIAM database annotations (http://www.ebi.ac.uk/miriam/main/) and notes. To add an annotation, press "Add annotation". A row will be added to the annotation table. Select the desired MIRIAM relationship (http://www.ebi.ac.uk/miriam/) and the database. Enter an identifer on the last cell of the row. To add a note, press "Edit notes" to obtain an "Edit notes" dialog. Edit the HTML code as desired, and press "Done." To delete an annotation, select a cell in the row of the annotation to delete, and press "Delete selected".
To update MIRIAM annotation options, beyond those contained within the program, the user may write text files in his/her home directory that contain updated selections.
To add annotation databases, create a file called "miriam.text" and save additions in the required format. Format examples are:
Aclame2==urn:miriam:aclame==http://aclame.ulb.ac.be/perl/Aclame/Genomes/mge_view.cgi?mode=info&id=$id
ArrayExpress2==urn:miriam:arrayexpress==http://www.ebi.ac.uk/arrayexpress/experiments/$id
arXiv2==urn:miriam:arxiv==http://arxiv.org/abs/$id
where the terms beginning with "urn" are MIRIAM universal resource identifiers (http://www.ebi.ac.uk/miriam/). To update relationship types, create a file called "relationelements.txt" in the user's home directory. Some example test relationships and their format for the file are:- bqbiol:encodes2
- bqbiol:hasPart2
- bqbiol:hasVersion2
- bqmodel:isEntire
Note that double-clicking on a row of the annotation table in a dialog causes the user's browser to access the associated URL contained within the program.
- Many elements within level 3 models have element histories. Some non-model elements in level 3 have element histories. In contrast with automatic addition of creation and modification dates in the model history, these history elements allow the user to set the dates of creation and modification, as well as list creators. This information is set when the user enters information in the dialogs and then presses "Done".
- The function definition dialog contains a separate field for arguments to the body of the function definition. A series of arguments must be entered as a series of arguments separated by a comma.
- Units and the UnitDefinition dialog: Units in an SBML model are referred to as UnitDefinitions. Each UnitDefinition is composed of one or more Units. A UnitDefinition is constructed in an "algebraic" manner, in which the product of several Units (unit1 times unit2 times unit3...) comprises a UnitDefinition.. UnitDefinitions are constructed using the Unit Definitions dialog. Press "New Unit definition" to bring up a new UnitDefinition panel. Press "New unit" to add a new Unit panel for building a new Unit for the the UnitDefinition that is showing. Once saved, UnitDefinition IDs may be used in several locations in SBML dialogs.
- Rule dialog: To create a rule, first select a rule type. If an Assignment or a Rate rule is selected, the referenced entity type and variable field will become enabled. The math expression applies to the variable that is selected. Choose a referenced entity type and select the desired variable.
Writing an SBML model
Ensure that the desired SBML level and version are set on the SBML Model dialog when beginning to design a model. Enter model content using the SBML dialogs. The SBML Default save dialog, available on the SBML menu, allows the user to turn off or on automated species ID and compartment creation, as desired. Using the Save SBML dialog available on the File menu, save the model. JSBML does not validate models against SBML model specifications. To check whether a model is in accordance with SBML specification, use the on-line validator at http://sbml.org/Facilities/Validator/. Local validation is expected to be added in the future. The "Clear SBML model" option on the File menu clears the SBML model.
MathML
Math expressions in SBML are written using a subset of Mathematics Markup Language (MathML). Please consult http://www.w3.org/Math/Software/ and SBML specification documents at http://www.sbml.org for information. The Pathway Editor uses JSBML to convert MathML expressions to formula string or algebraic formats (such as x * y, which represents the product of 2 variables x and y). The format is the same as that specified in SBML level 1. Unfortunately, later SBML levels are more advanced and the MathML may not convert to formula strings. Thus, by default, math expressions must be entered into models in MathML format. Also, because the Pathway Editor makes use of a pre-release version of JSBML, the Pathway Editor does not currently enable reading, editing, or writing SBML models that make use of SBML constructs within MathML called "csymbols". Csymbols represent, for example, simulation time, delay time, or Avogadro's number. The user should find alternate ways of expressing these in a model.
To enter and save formulas in MathML:
- Make sure the button above the Math expression entry area shows "Show formula".
- Type or paste the MathML containing the text into the area.
- Enter other information into the dialog as desired.
- Press "Done" on Windows or close the dialog on Macs. The program will check each MathML entry for validity and will report any errors.
- If all entries are valid, the dialog will accept the entries and will disappear. Note, however, that SBML contains many rules that are not checked by JSBML. The on-line validator is a resource that should be utilized for more thorough error checking.
To enter and save formulas in formula string or algebraic form:
- Make sure that the button above the MathML entry area shows "Show MathML".
- Type or paste the MathML containing the formula string in the tab.
- Enter other information into the dialog as desired.
- Press "Done" or close the dialog on Macs. The program will check each MathML entry for validity and will report any errors.
- If all entries are valid, the dialog will accept the entries and will disappear. Note, however, that SBML contains many rules that are not checked by JSBML. The on-line validator is a resource that should be utilized for more thorough error checking.
The button above the MathML entry area can be used to toggle between formula string and MathML views. However, if the area contains a csymbol, or more advanced MathML elements, some of the content may be lost. This is because of difficulty translating between MathML and formula strings.
BioPAX Support
Introduction
The BioPAX ("Biological Pathway Exchange", http://www.biopax.org) model file format provides for descriptions of pathways in text form. The Pathway Editor supports levels 2 and 3. Within the Pathway Editor, BioPAX models may be read from file, modified or constructed from scratch, and saved as either a .path file, for further editing within the Pathway Editor, or in .owl or .xml format for further editing in other applications.
BioPAX model files are structurally more complex than the basic Pathway Editor file format. Following is a brief description of the relationship between BioPAX and Pathway Editor requirements. These instructions address primarily level 2. Level 3 usage is analogous.
In BioPAX, the basic entity is a physical entity, analogous to a compound in the Pathway Editor path layout. The participants of an interaction are formally defined as Interaction Participants. In the Pathway Editor, participant nodes are not required to contain a compound. However, BioPAX requires that each Interaction Participant contain a physical entity.
One or more BioPAX interactions comprised of Interaction Participants may be represented by a single Pathway Editor interaction (for example, a biochemical conversion plus a catalysis). In BioPAX, additional links may be defined between different pathways and between pathways and nodes contained within other pathways. BioPAX links between components of pathways are not shown directly on the Pathway Editor drawing panel, but instead are shown in dialogs.
Many features of BioPAX are optional, such as entities referred to as pathway steps. Pathway steps may contain complete pathways and nested subpathways, or may just contain a series of interactions, or combinations of subpathways and interactions.
The Pathway Editor uses identifiers of entities given in BioPAX files that are read by the program, and will also assign identifiers to new entities that are added by the user. Identifiers for BioPAX entities are not shown in the Pathway Editor, but instead are managed out of view of the user.
BioPAX model files
- BioPAX files are read into the Pathway Editor by selecting the "Open BioPAX pathway file" item on the File menu. File names ending with ".owl" or ".xml" are allowed.
- After a BioPAX file is opened, pathways, subpathways, interaction participants, and their physical entities are listed on the BioPAX Reader dialog. Interaction participants and sub-subpathways contained within enclosing pathways are shown on tabs for the enclosing pathway.
- Individual pathways and participants may be selected and transferred to the drawing panel one at a time. To achieve this, select a checkbox and press "Center node". Pathway interactions are automatically drawn, and a check mark is added to the selected row. Information on the drawn entity may be obtained by right-clicking on the drawn node and selecting from the pop up menu.
- As an alternative to individual transfer of interaction participants, the "Transfer all" button transfers the entire contents of the model. If the "Auto layout" radio button is also selected, the program lays out each pathway to separate the nodes. Move the nodes around to a preferred layout. The layout is preserved when the model is saved as a .path file. The .path file can be used in conjunction with the BioPAX model file.
- Other controls on the BioPAX Reader allow for transferring BioPAX complexes as Pathway Editor nodes of different types, and for selecting the node label that will be displayed. A node label is required before the node can be drawn.
- To reverse the transfer process and return to a clear drawing panel, press "Reset" on the Reader. However, if a change is made to the model, the reader dialog will be cleared automatically.
- To reuse a saved .path file that has been written after constructing or reading a BioPAX model file, first read in the BioPAX file, but do not transfer to the drawing panel. Then read in the .path file. BioPAX entities are automatically assigned to each node.
Building an example BioPAX model
- Draw two small molecule nodes and connect them. Then add a protein node and connect to the interaction between the small molecules. Assign labels "SM1", "SM2", and "Catalyst."
- One at a time, right-click on each node and select "BioPAX entity info." A BioPAX small molecule dialog appears for the small molecules, while a BioPAX protein dialog appears for the protein.
- Enter information on the BioPAX physical entities, for example, by naming them "sm1", "sm2", and "catalyst." Press the "Done" button to save. Note that it is not necessary to enter anything in the dialog in order to create a BioPAX physical entity — in such a case, the text fields will remain empty in subsequent showings. To see if a node has a BioPAX physical entity, go to "BioPAX | Physical entity participants" to view a list of nodes and physical entities. The physical entity name for a node that has not been assigned an entity will be "unassigned," while the name listed for a node that has been assigned an entity but that does not have a name will be "NO_NAME."
- Right-click on the interaction and select "Show info". On the Interaction dialog that appears, select the "BioPAX pathway step" tab.
- Press the "Add new interaction" button.
- Using the "Create new BioPAX interactions" dialog, create a BioPAX biochemical reaction. Select SM1 and press the "Select" button to move SM1 from the left to the right-hand list. Then select SM2 and press the "Select" button to move SM2. Select "Biochemical reaction" on the "Type" drop-down list at the top of the dialog. Press "Create." A biochemical reaction will be created, with the names of the nodes and the interaction type displayed on the "BioPAX pathway step" tab. The "Create new BioPAX interactions" dialog will reset.
- Create a catalysis interaction: On the "Create new BioPAX interactions" dialog, select the "Catalyst" name and press the "Select" button. Select "Catalysis" on the "Type" list and press "Create". A catalysis interaction will be created and a second row will be entered on the BioPAX pathway step tab. Press "Done" on the "Create new BioPAX interactions" dialog.
- On the BioPAX pathway step tab, select the row for the catalysis and press the "Show interaction" button. A BioPAX catalysis dialog will appear. On the Catalysis tab, press the "Select controller" button. In the "Select controller node" dialog, select "Catalyst" and press the "Add to list" button. Then press the "Done" button. On the Catalysis tab, the Catalyst name will appear in the Controller text field.
- On the Catalyst tab, press the "Select controlled" button. The "Select controlled reaction" dialog will appear. Select "Biochemical reaction(SM1, SM2)" and press "Add to list." Then press the "Done" button. On the Catalysis tab, the reaction will appear in the Controlled text field. Press "Done" to return to the BioPAX pathway step tab. The tab will contain a new row with the node "Catalyst" and the interaction type "Catalysis".
- If desired, the set of reactions can be added to a pathway step. Press the "Manage step" button.
- The "Manage pathway steps" dialog will appear. To create a new step, press the "Create step" button. The name of a new pathway step will appear in the list. Select the step in the list and press "Add to list." Press "Done". The dialog will disappear and the name of the step ("Step 1") will appear on the "BioPAX pathway step" tab.
- Create a top-level pathway entity by selecting the menu item "BioPAX | Pathways." The "Manage BioPAX pathway" dialog will appear. Press the "Create top pathway" button. The title of the tab will change from "No pathway" to "Top pathway." The attributes of the pathway can be set as well. On the "Pathway components" tab, "Step 1" will appear. To also see the BioPAX interactions, press "Step interactions." The BioPAX interactions will be shown in the list as "Biochemical reaction(sm1, sm2)" and "Catalysis(Catalyst protein, Biochemical reaction(sm1, sm2)). The properties of each step or interaction can be viewed by selecting each and pressing "Show component."
- Note that selecting a component on the "Pathway component" tabs of BioPAX pathway dialogs causes the component to be outlined on the drawing panel, provided the component is showing.
- To save the model as a .owl file, go to "File | Save BioPAX pathway file" and save using the resulting File save dialog. To preserve the layout, save it as a .path file using "File | Save pathway as."
BioPAX Interaction Participants
- In BioPAX, interactions are composed of Interaction Participants. Every Interaction Participant must have a unique physical entity. There is no sharing of physical entities. Before a BioPAX entity can be added to a BioPAX interaction, the BioPAX entity must first be defined (small molecule, protein, DNA, RNA, or complex). The Pathway Editor manages BioPAX Interaction Participants automatically, beginning when the user creates a BioPAX interaction.
- Pathways can be Interaction Participants only in physical interactions.
- Stand-alone physical entities and Interaction Participants that may be contained in a BioPAX model file are not recognized by the Pathway Editor. Thus, only Interaction Participants that actually participate in interactions are read by the program.
- Interaction dialogs for BioPAX interactions show the properties of Interaction Participants on separate tabs (except for pathways).
- Unfortunately, Interaction Participants cannot be added to an existing BioPAX interaction. It is necessary to delete the old BioPAX interaction and then create a new one containing the desired Interaction Participants
BioPAX interactions
- A Pathway Editor interaction may consist of one or more BioPAX interactions. For example, a combination of BioPAX interactions may join at a Pathway Editor interaction icon that represents the aggregation of a BioPAX biochemical reaction and a BioPAX catalysis.
- BioPAX interactions are created after Pathway Editor interactions are created and BioPAX participants are assigned to nodes. They are created in a series of steps that starts by pressing the "Add interaction" button on the BioPAX tab of Pathway Editor interaction dialogs. A metabolic pathway of biochemical reactions that are catalyzed by catalytic molecules may be built up by first creating a biochemical reaction, then creating a catalysis on the same BioPAX tab. Other combinations of BioPAX interactions are possible.
- If BioPAX interactions for a Pathway Editor interaction are displayed in a Pathway Editor interaction dialog, the Pathway list on the BioPAX tab shows the BioPAX pathway that contains the interactions. This list is editable, but changes made here are not effective. To insert BioPAX interactions into a pathway, two options are available: (a) expand a subpathway node and modify or draw the desired reactions and construct BioPAX interactions, and then compress the subpathway; and (b) draw or modify interactions on the panel, and then insert into a top-level pathway as in the tutorial above.
- BioPAX interaction dialogs may be viewed by selecting the "BioPAX entity info" option after right-clicking on a Pathway Editor interaction icon. BioPAX interaction dialogs may also be viewed from other BioPAX dialogs, and from several Pathway Editor dialogs.
- To edit the directions of BioPAX interactions (that is, left or right), use the node properties tab of the Workbench interaction dialog.
- BioPAX interactions may be constructed between components that are internal to pathways and external objects. An interaction must first be created using the Pathway Editor (see the "Subpathway interactions" section). The BioPAX interactions are then created. Neither kind of interaction is shown on the drawing panel, but is seen only in dialogs. A convention that may be useful in some situations is to construct connections between pathway nodes on the drawing panel, without actually adding BioPAX to the connections, to suggest the presence of BioPAX interactions.
BioPAX pathway steps
Pathway steps are optional entities that enclose one or more BioPAX pathways, pathway steps, and interactions. Pathway steps may be created by the user after BioPAX interactions are created, using the "Pathway steps" dialog obtained from the BioPAX interaction tab of interaction dialogs. Lists of pathway steps are shown on the "Pathway steps" dialog, obtained by selecting the BioPAX | Pathway step menu. Select from the list at the top of the dialog to see the properties of individual steps. The comments tab shows comments for the pathway step that is selected.
- BioPAX pathway steps may be created on the BioPAX tab of Pathway Editor interaction dialogs. Press the "Manage step" button. On the "Manage pathway steps" dialog, select one of the steps in that already exist, or press the "Create step" button to create a new step. Press "Add to List". The newly created step will be added to the list on the "Manage pathway steps" dialog. Press the "Done" button. The name of the step will be transferred to the BioPAX tab and the list of one or more BioPAX interactions will be part of the step.
- Pathway steps that enclose a pathway may also be created. Right-click on a pathway on the drawing panel and select "BioPAX entity info". On the "Manage subpathway" dialog, press the "Create/select step" button and utilize the "Manage step" button as in step 1.
- Note that pathway step names are not part of BioPAX. Names ("Step 1", "Step 2", and so on) are assigned by the Pathway Editor during step creation and when a BioPAX model file is read.
- A list of pathway steps and interactions within a pathway appears on the "Pathway components" tab when the BioPAX pathway dialog is subsequently displayed. Pathways steps and interactions are outlined on the drawing panel when the corresponding name is selected.
- The order of pathway steps may be set on the "Pathway steps" dialog. Press the button "Set order." The "Next steps selection dialog" will appear. Select a step on the lower list and press "Add to list." The step will be transferred to the upper list. More than one step may be set. When finished, press "Done." The dialog will disappear and the selected steps will appear on the "Next steps" list of the "Pathway steps" dialog. To view the next step on the "Pathway steps" dialog, select one of the next steps, and press "Show next."
BioPAX pathways
BioPAX pathway entities are optional in a BioPAX model. If a pathway is present, it is shown as a subpathway node on the drawing panel. The attributes of pathways are set on the dialog that is obtained by right-clicking on the pathway node and selecting "BioPAX entity info." Attributes can also be set on the "Manage BioPAX pathways" dialog, obtained by selecting the BioPAX | Pathway menu.
A single top-level pathway that encloses all other BioPAX entities can be established using the "Create top pathway" button on the Manage BioPAX pathways dialog. At least one BioPAX interaction and two nodes containing BioPAX Interaction Participants must be showing on the viewing panel. The presence of a tab containing the title "Top pathway: Top pathway" indicates that a top-level pathway has been created. The top pathway can be removed using the "Remove top pathway" button.
A new BioPAX pathway may be created by first constructing a subpathway node. Then right-click on the subpathway and select "BioPAX entity info." Enter the attributes, synonyms, and comments to be assigned to the new pathway. Many attributes, such as the biosource, datasources, and so on must be created first using other BioPAX dialogs. When finished, press "Done" to create the pathway and insert all the sub-entities contained within the compressed node.
BioPAX utility classes
BioPAX requires that certain kinds of information, such as chemical structures, references to external sources, and publication and database references, be defined separately from the participants in interactions. This information may be entered or viewed in dialogs available from the BioPAX menu. If needed, Pathway Editor names signifying a utility information may be obtained from utility class dialogs and entered into other BioPAX dialogs as necessary, using selection dialogs called by pressing buttons. Because the designers of BioPAX intend that BioPAX identification tags in a model not be shown to users, the Pathway Editor creates names that can be used to track this information for presentation purposes. Thus, in the process of model building, these names may change each time the model is read from file. See the BioPAX documentation for details.
CellML Support
The CellML model file format
The Pathway Editor reads, constructs and edits mathematical models in CellML. Both CellML 1.0 and CellML 1.1 are supported (http://www.cellml.org/specifications).
In CellML, reactions (reactants, participants, and enzymes) are not supported. The actual term is "deprecated", meaning that though the specification documents describe reactions, their implementation is discouraged (http://www.cellml.org/getting-started/cellml-primer#section-7)). Mathematical expressions in CellML files are described using MathML (www.w3.org/Math/). CellML does not specify visual presentations, except for presentation of mathematical equations. However, there is no agreed upon mechanism for visually editing either CellML or MathML. Consequently, nearly all of the manipulation of CellML and MathML content is performed using text entered into dialogs.
After a specification version is set on the Model dialog, the version cannot be changed.
- Installing the Pathway Editor that incorporates CellML
The Pathway Editor uses code libraries that are specific to each operating system. Unfortunately, they are not accessible from Java Web Start. Consequently, a stand-alone program must be downloaded and installed for this functionality. For CellML in a stand-alone program, download and install a separate installer program using one of the following links. These installers were created using Install4j:
Mac- PathwayEditor_macos_1.0.dmg (separate Java Runtime Environment not required)
- PathwayEditor_macos_nodb_1_0.dmg (no database connection) (separate JRE not required)
Mac OS X 10.5: CellML code libraries do not work on this version. Please use the Web Start version (no CellML).
On Mac platforms, double-click on the .dmg file on the desktop or in a Finder dialog. Then, double-click on the Pathway Editor insta ller icon or row in a Finder dialog to install. After installation, double-click on the Pathway Editor icon or row in the Finder dia log.
Windows- PathwayEditor_windows_1_0.exe (separate JRE not required)
- PathwayEditor_windows_nodb_1_0.exe (no database connection) (separate JRE not requred)
To install on Window platforms, double-click on the Windows executable and follow the dialogs. After installing on Windows, the option to start the Pathway Editor will appear on the list of installed programs that are accessed from the list of program files and also from the Start menu. For platforms other than the above, please contact the webmaster.. After installing on Windows, the option to start the Pathway Editor will appear on the list of installed programs that are accessed from the list of program files and also from the Start menu.
For platforms other than the above, please contact the webmaster.
- CellML dialogs in the Pathway Editor
Dialogs in the Pathway Editor allow inspection of model content by selecting names in tables. Generally, selecting a row of a table will lead to tables underneath the selection table to become populated with information relevant to the selected element and its sub-elements. This convention is used because of the generally hierarchical nature of CellML. Buttons associated with these tables become activated when such selections are performed.
The CellML file specification contains reactions; however, the website of the CellML project (www.cellml.org) discourages their use (http://www.cellml.org/getting-started/tutorials/the-now-defunct-reaction-element-in-cellml/?searchterm=deprecated).
Reading and writing CellML model files, and clearing models, is performed using menu items on the File menu. CellML dialogs are accessed from the CellML menu.
- Opening a CellML model file: The CellMLReader
The CellMLReader dialog is displayed when a CellML model file (name ending in .cellml) is read using a dialog selected on the File menu, or the "CellML reader dialog" is selected on the View menu.
The CellMLReaderDialog dialog works similarly to the SBMLReaderDialog and the BioPAXReaderDialog, except that interactions are not drawn when transferring nodes to the drawing panel.
As a substitute for reaction components, the CellMLReaderDialog transfers components and variables to the viewing panel but does not draw interactions. CellML components are represented by subpathway nodes, while variables are represented by the usual nodes for different compound types. Thus, variables serve as placeholders for reaction participants. Because there are typically more variables than needed for representation of reactions, the user needs to decide which variables are worthy of being part of a pathway drawing.
- The user also needs to decide whether or not to display components from CellML models as subpathways, possibly hiding any reactions within subpathway nodes that are assigned to represent components.
- The "Transfer all tabs" button draws components as subpathways and includes all variables as nodes of the selected type.
- A suggested technique is to select nodes on one or more tabs to be laid out, and press "Transfer all selected". Only components and the rows that are selected on each tab will be transferred. Alternatively, press "Transfer all tabs". After this action, on the drawing panel, expand subpathways one at a time and delete undesired nodes.
- After or during the above, draw any desired reactions.
- To preserve the layout, save the pathway as a Workbench .path file to be read into the program after a .cellml file is first read but before variables are laid out.
- The Model dialog
The model element is the highest level structure in CellML. In actuality, the �Document� element may perhaps be a higher-level element, as a document may be considered to contain a model plus metadata that is associated with the document. A document element is not explicitly indicated in the CellML file, but it is referenced in the Metadata Dialog only for purposes of tracking metadata.
The Model dialog shows several kinds of model constituents that are defined or constructed using their own dialogs.
Every model must have a name. Models may contain units that may be used by components. These components may be comprised of model components as well as components that are imported. Imports are composed of model units and components that are extracted from other models and may be used to add computational features. This is an example of reuse.
Units are shown in the Units table when a component is selected. The "Model units" button causes units that are defined within models to be listed in the Units table.
- The CellMLComponent dialog
Components are the smallest functional unit of a model. A single component can represent a valid CellML model (Lloyd et al, Prog. Biophys. Molec. Biol 85 (2004), 433-450). They contain variables and one or more mathematical formulae that describe biological processes. They may also contain units that are used locally by the component.
The CellMLComponent dialog shows names of components within the model, as well as any imported components.
Units may be defined within a component, but the units are not made available for use outside the component. Variables may be assigned both local units and model units.
Formulas in components are constructed in XML in the form of MathML elements and are listed in the MathML table as "Expression 1", "Expression 2", and so on.
- The Units dialog
The Units dialog may be used to build units that are associated with variables inside components. Units may also be placed inside a model element, in which case they are shared among components. Units may be either standard units that are a part of CellML, or they may be defined by each model.
The top table shows a list consisting of the model name, imports, components, and imported components. Selecting any of these rows causes the "Units" table to become populated with a list of the units within that element.
Selecting a "Units" in the Units table will show the structure of the Units in the form of separate "unit" elements on tabs of the tabbed pane at the bottom of the dialog. These unit elements comprise the blocks upon which units elements are constructed. The properties of each unit may be set. After setting unit properties, "Accept tabs" must be pressed, after which "Done" must be pressed to save the contents of the entries.
- The Variables dialog
The Variables dialog defines variables that are used within components. Variables consist of a name and a "units", that is in turn composed of 1 or more "unit" elements. Variables may be assigned a standard units that is part of the CellML language, or variables may have units that are user-defined.
The top table contains lists of component and imported component names. Selecting one causes the "Variables" table to become populated.
In turn, selecting a variable name causes the controls beneath the Variables table to display the properties of the variable.
The "Public interface" and "Private interface" properties allow grouping of components in different relationships with respect to each other. These relationships allow sophisticated modeling calculations to be performed independently when used within a simulation program.
- Connections
Connections loosely defined are interactions between 2 components, in which values of a variable may be obtained from 1 component and used in a second component during simulations.
The Connections dialog allows definition of connections. Connections may be constructed between components of a model, and between model components and imported components. There are restrictions on constructing connections, based upon relationships between groups.
The CellML specifications contain explanations of the importance of connections for modeling purposes. See the section on groups found below for more information.
On first opening the Connections dialog, a list of all connections in the model appears at the top in the "Current connections" table. If one of the listed connections is selected, details of the connection are listed in the tables below. The "Current variables" tables show the variables and units that currently comprise the connection.
To construct a new connection:
- Press "Reset".
- The "Candidate variables" tables show all variables in the two components that are available for connections between these two components, according to specification rules.
- Select candidate variables, based upon common units and their details on the Variables dialog. Press "Create connection". After successful addition, the candidate variables will be shown in the "Current variables" tables.
- Pairs of candidate variables may be added to an existing connection. Individual pairs of current variables, however, cannot be deleted. In this case, the entire connection should be deleted and a new connection created, leaving out the undesired variables.
- To delete a connection, select a connection and press "Delete connection". The connection will be removed from the model and the revised list of connections will be shown.
- Groups and relationships
Groups are assemblies of components that restrict connections according to relationships. CellML specifications define two relationships that are named "encapsulation" and "containment". For a description of restrictions imposed by encapsulation, please consult the CellML specifications. Containment is also a relationship that is named in the specifications, but restrictions are stated to be imposed by the processing software rather than the CellML software itself. The Pathway Editor allows use of containment, but it does not associate restrictions with containment.
In addition to encapsulation and containment, any arbitrary relationship may be added to the model. The rules for grouping of these relationships are enforced by processing software outside the Pathway Editor.
When the Group dialog is first displayed, all groups defined by the model are shown in the "Groups" table. Selecting one of the groups causes the relationships for the group to be shown in the "Relationships" table. The "Component hierarchy" table is also filled. Top-level components are shown beginning at the left side of the table. Sub elements of the hiearchy are indented.
To create a new group:
- Press "Add". A new group name will appear in the Groups table.
- Select the new group name and press "Edit". The Relationships dialog will appear. Select checkboxes for one or both of the two defined relationship types as desired. Alternatively or in addition, a new relationship can be defined by pressing "Add". A new row appears in the Relationships dialog. Enter the new type and an optional namespace. Press "Done" to cause the dialog to disappear and transfer the descriptions to the Relationships table.
- Select the new relationship in the relationship table. To add a top-level component, press "Add top components". A selection dialog will appear. Select one or more of the components and press "Add to list".
- Press "Done". The selection dialog will disappear and the top-level components will be listed on the groups dialog.
To add a subcomponent:
- To add a subcomponent to a top-level component, select a top-level component and press "Add subcomponents". Use the selection dialog in the same fashion as in c and d above.
To replace a component:
- To replace a component in the hierarchy, select the component to be replaced and press "Replace component". A dialog to select the replacement component will appear. Select a component and press "Done".
To delete a component:
- To delete a subcomponent, or a top component and any of its subcomponents, select the component and press "Delete".
- The MathML dialog
The MathML dialog allows management of formulas (expressions) in MathML (XML) format. When the MathML dialog first appears, components and imported components are shown in the top table. Expressions take the general form of two-sided equations such as y = f(x).
- Select a component in the Component table. The Math expressions table becomes populated with a list of the expressions within the component.
- Select one of the expressions. The MathML will be displayed in the "Expression" area.
- After selecting the expression, the MathML may be changed by the user by typing into the Expression area. Press "Update expression" to accept the update.
- To add a new expression, first press "Reset" to clear the expression area. Select a component. Enter new MathML into the area. Press "Add expression" to add it to the component.
- To remove a selected expression from the component, press "Delete expression".
- Imports
CellML models that conform to the CellML 1.1 specification may be combined by importing model units and components from one file into a second. Once imported, imports cannot be modified.
For purposes of constructing connections, the units of an imported component must be defined in the imported model, not in the imported component.
In the Import dialog, the component name and units name tables contain the names that may be used in the current model as a substitute for names in the imported model in the first column. This permits avoidance of name clashes.
Connections can be made to imported components, and imported model units can be used within existing components. Variables, component units, and MathML in imported components can be inspected but cannot be modified using dialogs on the CellML menu.
In some cases, a component in an import must access other components in the import, which are part of groups. Consequently, these other components are imported automatically into the current model.
To import from a model:
- Open the Imports dialog from the CellMLMenu.
- Press "Add imports(s)".
- In the file open dialog, navigate to the desired model and select it.
- An import selection dialog will appear. Select components and model units.
- Press "Done" on the import selection dialog.
- The import will be shown as "Import 1", "Import 2", etc. in the top table of the Import dialog, and the components and units that have been imported will be shown in the tables.
- Components and units can be deleted or renamed for local use on the import dialog. Pressing "Clear all" will remove all imports from the model.
- The Metadata Manager and Citation dialogs
The Pathway Editor can display, modify, and add metadata, or information about a CellML model, according to the CellML Metadata 1.0 specification. The metadata is contained within "Description" elements. Any CellML element may be described by one or more Description elements.
CellML model files may contain other kinds of metadata than described in the CellML Metadata 1.0 specification. Such metadata is not displayed.
The Metadata Manager dialog is displayed when the "Metadata manager" menu item on the CellML menu is selected.- The "Element types" table contains a list of CellML element types. Select the desired type with your mouse.
- The "Element" table will show the elements of this element type. Each element that contains a Description will be followed by an asterisk.
- Select the desired element in the Element table.
- If the selected element contains metadata, the "Metadata descriptions" and the "Citations" table will contain entries. Note that citations or bibliographic references also appear in descriptions, but have a separate dialog for displaying.
- If selections are made in these last two tables, pressing the "View desc" or "View citation" buttons will show a "Metadata dialog" or a "Citation dialog", respectively.
- These last 2 dialogs allow inspection and editing of metadata and citation features separately.
- A new Description or a new citation may be added to each element using the "Add metadata" or "Add citation" button next to the element table. Select the desired element and press the desired button.
- Descriptions or citations may be deleted by selecting in the "Metadata descriptions" or "Citations" tables and using the respective delete button. If a description that contains only a citation, deleting the citation will also delete the description.
- Note that selecting a description that contains only a Bibliographic reference will have an entry in the Descriptions table, but will show a blank Metadata dialog.
- Book articles are a special case. The Citation dialog for a book article shows information immediately pertinent for the article. Because book articles are contained within a book, additional citation information may be relevant to the book itself. Pressing �Book details� on the Book article tab will bring up a second citation dialog in which information on the book may be entered.
- Technical note on citations in CellML: Because of loose terminology in the CellML Metadata 1.0 specification, some Description elements contain a citation within another citation. The top reference is a "Reference" element. The sub citation is usually a more specific type of citation, such as a journal article. The Pathway Editor recognizes this kind of structure. In fact, this is how the Pathway Editor creates new references. However, an XML element structure in which a citation does not contain another citation is also recognized and is not changed when edited in the Pathway Editor.
- On the SBML model creation dialog, select "Save pathway layout" if the Workbench pathway layout is to be saved as an annotation.
- The default compartment ID is c1; this can be changed after model creation in the Compartment dialog.
- Select whether modifier species references are to be created from proteins.
- Press "Create now".
- Species, species references, and reactions are generated. Species annotations contain database annotations.
- On the BioPAX options dialog, select whether catalyst interaction controllers are to be created from proteins. This selection also enables creation of catalysis reactions.
- Select whether DNA or RNA objects are to be created from nucleic acids.
- Press "Create now".
- Biochemical reactions and catalysis interactions are generated. Subpathway nodes are converted to BioPAX pathways. Links between participant nodes within subpathways are also preserved as Interaction Participants. Compound synonyms are transferred into synonyms for each BioPAX small molecule, protein, and nucleic acid. Unifications are created from database references and become associated with these BioPAX elements as well.
- On the CellML settings dialog, press "Create now".
- A single component "Component1" is created, with variables that take the names of nodes. Compound synonyms are transferred into Descriptions as alternative element titles or names. The biological entity panels of metadata description dialogs for variables contain database references for the compound.
- In IE, go to http://java.com/en/download/ie_manual.jsp?locale=en. Press the red download button and begin installation. If you get a dialog that says something like "Java is already installed", ignore it. As of this writing, the latest version is Java 6, update 31.
- Allow the computer to restart when prompted.
- Navigate to http://www.lipidmaps.org/pathways/pathwayeditor.html and click on "Download and launch Pathway Editor".
- You will get a dialog asking you to set the program that opens the .jnlp file. Navigate to the most recent Java installation, such as C:\Program Files\Java\jre6\bin. Depending upon the download, "Java" and "jre6" may be replaced by directory names containing the version and update numbers for Java. Select javaws or javaws.exe (for Java Web Start).
- The Pathway Editor should start up.
- Go to Tools | Options at the top of the browser window. A dialog should appear containg a number of tabs. Select the "Applications" tab.
- The tab shows a table of file types under the column "Content Type", and the programs that are used to view the file type under "Action".
- Scroll down to Content type "JNLP file". On the right, click the cell "Use Java(TM) Web Start Launcher".
- The cell becomes a drop-down list, with 5 or more options. If you select "Application Details", you will see a dialog with the name of the program and its full name, containing a directory and the program name. A program called "javaws.exe" should be listed. If you wish, press "Remove". Note that each version of Java may have an entry in the list.
- If you select "Use other", another dialog will appear. At the top is "JNLP file". Underneath are the words "Send this item to:", and a list of programs.
- At the bottom of the last dialog are 3 buttons. Press the "Browse" button. You will see a dialog titled "Select Helper Application".
- In the "Select Helper Application", browse to the a directory containing javaws.exe and select the version of the javaws.exe that you wish to use. An example might be located at "C:\Program Files\Java\jre6\bin\javaws.exe". If you have more than one version of Java, the directory names Java and jre6 might contain the version and update number. You should choose the version with the greatest set of numbers, as this is the most recent.
- In the "Select Helper Application" dialog, press "Open". There should be a new item added to the Options table, titled "Use Java(TM) Web Start Launcher".
- Press "Close" to exit the Options dialog.
Annotations
If a BioPAX, CellML, or SBML model has been created, and a corresponding entity (physical entity participant, variable, or species) in one of these models has been associated with a node, annotations in these models are readily added. This is achieved by downloading a compound with its database references. The database references automatically become associated with the entity.
Creation of SBML, BioPAX, and CellML models from Workbench .path files
Creation of SBML, BioPAX, and CellML models from Workbench .path files
Basic SBML, BioPAX, and CellML models may be created from pathways laid out on the drawing panel. Existing compound database annotations (references) are transferred into the resulting models. After creating models, names may be modified and model type-specific details such as mathematical formulas may be added.
SBML
BioPAX
CellML
Troubleshooting
Pathway Editor will not open
Windows / Internet Explorer
Windows operating systems may update themselves automatically. In so doing, Internet Explorer sometimes loses track of the program that is used to open .jnlp files. These files are read by browsers when the user clicks on hyperlinks to the files. The browser passes .jnlp files to Java Web Start for downloading and processing of programs specified in the .jnlp file.
The following steps will ensure that Internet Explorer will remember to use Java Web Start on .jnlp files.
Mozilla Firefox and other Windows browsers
As a first step, you might try installing Java using your preferred browser, following the instructions for Internet Explorer according to the previous section.
Alternatively, or in addition to, a new installation of Java, a browser option can be set to use a different installation. This may be required when a Web Start program requires a more recent version of Java. The following instructions refer to Mozilla Firefox version 11.0. Other versions and browsers should have similar instructions.